

Abstract
A number of viral gene products are capable of triggering apoptotic cell death through interfering with cellular signaling cascades, including the Akt kinase pathway. In this study, the pro-apoptotic role of the SARS-CoV Membrane (M) structural protein is described. We found that the SARS-CoV M protein induced apoptosis in both HEK293T cells and transgenic Drosophila. We further showed that M protein-induced apoptosis involved mitochondrial release of cytochrome c protein, and could be suppressed by caspase inhibitors. Over-expression of M caused a dominant rough-eye phenotype in adult Drosophila. By performing a forward genetic modifier screen, we identified phosphoinositide-dependent kinase-1 (PDK-1) as a dominant suppressor of M-induced apoptotic cell death. Both PDK-1 and Akt kinases play essential roles in the cell survival signaling pathway. Altogether, our data show that SARS-CoV M protein induces apoptosis through the modulation of the cellular Akt pro-survival pathway and mitochondrial cytochrome c release.
Keywords: Cytochrome c, Drosophila, HEK293T, PDK-1, Severe Acute Respiratory Syndrome
In 2003, the Severe Acute Respiratory Syndrome-Coronavirus (SARS-CoV) emerged and caused an outbreak of atypical pneumonia worldwide. The SARS-CoV genome contains 13–15 open reading frames (ORFs) [1], [2], [3] which encode the ORF1a–1b (replicases and protease enzymes), Spike (S), Envelope (E), Membrane (M), Nucleocapsid (N), and a number of less well-characterized regulatory proteins [1], [2]. Previous studies revealed that SARS-CoV infection triggers a number of cellular responses in infected cells including modulation of signal transduction cascades such as the p38 MAPK and Akt cell survival pathways [4], [5], [6], [7], [8], [9], [10], [11], [12], and induction of apoptosis [11], [13], [14]. Apoptosis is a main pathologic feature induced by SARS-CoV infection [4], [11], [13], [14], [15], and a number of SARS-CoV proteins have been shown to be pro-apoptotic [12], [16], [17], [18], [19], [20], [21], [22]. Apoptosis induction has further been demonstrated to initiate viral cytopathic effect (CPE) 1 in the infected cells [13] but appears not to be a mechanism for dissemination of new virions [15]. Over-expression of individual viral proteins is now known to be capable of interfering with various cell signaling cascades. For example, the N protein can modulate the MAPK pathway and down-regulate 14-3-3 protein level [12], [23]. It is proposed that alteration of the status of various cellular signaling cascades, such as the Akt pathway, through the concerted effort of individual viral proteins would eventually deliver detrimental consequences to SARS-CoV infected cells [24].
The SARS-CoV Membrane locus is located in the 3′ region of the viral genome which encodes a protein of 221 amino acids [1], [2]. The SARS-CoV M protein is one of the major proteins among all viral gene products [25]. The M protein possesses a triple membrane-spanning region, an extracellular N-terminal and a long cytosolic C-terminal domains [26]. Physical interactions between M and other viral proteins, including N [27], [28], [29], S [30], 3a [31] and 7a [32], have been elucidated; and such protein–protein interaction events are predicted to be essential for the biological functions of M in the viral life cycle. Postulated roles of M include promoting membrane fusion, regulating viral replication, and packing genomic RNA into viral particles [26], [33]. Apart from the well-documented roles in viral infection and propagation, novel functions of various SARS-CoV structural proteins such as N and S have recently been assigned [12], [19], [20], [22]. In the present study, we aimed at investigating previously unidentified roles of the SARS-CoV M protein. We over-expressed the SARS-CoV M protein in cells and transgenic Drosophila, and observed that M was capable of inducing a mitochondrial-mediated caspase-dependent apoptosis. We also showed that M over-expression down-regulated Akt protein phosphorylation.
Materials and methods
Construction of mammalian expression vector
Full length ORF of the SARS-CoV Membrane locus was PCR amplified from viral cDNA template (CUHK-Su10 SARS-CoV isolate; GenBank Accession No. AY282752) and subcloned into pcDNA3.1(+) vector using EcoRI and XbaI enzymes to generate the pcDNA3.1(+)-Membrane construct.
Mammalian cell culture and transient transfection
Human embryonic kidney cell line HEK293T was maintained at 37 °C in Dulbecco’s modified Eagle’s medium (DMEM, Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum (Gibco-BRL), streptomycin (100 g/ml), and penicillin (100 U/ml). Cells were seeded onto 6- or 24-well plates 24 h prior to transfection. Four (6-well plate) or 0.8 (24-well plate) μg of plasmid DNA were used for transient transfection with Lipofectamine 2000 reagent (Invitrogen). Cells were collected for the subsequent analyses 48 h post-transfection.
Semi-quantitative RT-PCR analysis
Total RNA was prepared as previously described [34]. Primers used were GAPDH-F: 5′ ACC ACA GTC CAT GCC ATC AC 3′; GAPDH-R: 5′ TCC ACC ACC CTG TTG CTG TA 3′; M-F: 5′ ATT ACC GTT GAG GAG CTT AAA CAA 3′; and M-R: 5′ CAA TGA CAA GTT CAC TTT CC 3′.
Immunofluorescence staining of HEK293T cells
Cells were seeded onto poly-d-lysine-coated coverslips at a density of 2 × 104 cells/coverslip. After transfection, cells were fixed with 3.7% formaldehyde for 15 min and then permeabilized by 1% Triton X-100 for 5 min. After blocking with 1% goat serum for 30 min, cells were incubated with rabbit anti-SARS-virus-PUPM antibody (C-term) (1:100; Abgent) alone or in combination with mouse anti-native cytochrome c clone 6H2.B2 (1:100; Pharmingen) at 4 °C overnight, and followed by secondary antibody incubation with goat anti-rabbit IgG (H+L)−FITC (1:250; Zymed) alone or in combination with goat anti-mouse IgG (H+L)−TRITC (1:250; Zymed) at room temperature for 1 h. Golgi body was stained by BODIPY® TR ceramide complexed to BSA (5 μM, Molecular Probes) and cell nuclei were labeled with Hoechst 33342 (trihydrochloride trihydrate) (5 μM, Molecular Probes) at room temperature for 10 min. Cell permeable synthetic caspase inhibitors (Merck) Z-DQMD-fmk (caspase-3 inhibitor V), z-IETD-fmk (caspase-8 inhibitor II) and z-LEHD-fmk (caspase-9 inhibitor I) were dissolved in DMSO. HEK293 cells were treated with caspase inhibitors (50 μM in 1% DMSO) 24 h post-transfection, and were further incubated for another 24 h. Fluorescence images were captured using an Olympus BX51 upright fluorescence microscope or a Leica NT confocal microscope.
Drosophila genetics
Fly strains were grown at 29 °C on standard cornmeal medium supplemented with dry yeast. PDK-1 1 and PDK-1 2 lines were kind gifts of Jongkyeong Chung [35]; gmr-GAL4, UAS-DIAP1, UAS-P35, P[GMR-Akt1.Exel]2, Exelixis, and DrosDel lines were obtained from Bloomington Drosophila Stock Center; and the dc3 EP2305 and PDK-1 EP837 lines were obtained from Szeged Drosophila Stock Centre.
Generation of transgenic fly lines
Full length ORF of the SARS-CoV Membrane gene was PCR amplified from viral cDNA template and subcloned into pUAST vector [36] using EcoRI and XbaI enzymes to generate pUAST-Membrane plasmid. Standard microinjection technique was employed to generate transgenic lines. A total of 10 UAS-Membrane transgenic fly lines were generated. When crossed to gmr-GAL4, all lines showed dominant rough-eye phenotype. A transgenic fly line J3 carrying an UAS-Membrane insert on the third chromosome was used in this study.
Scanning electron microscopy of adult fly eyes
In brief, fly heads were fixed in 2.5% glutaraldehyde (EM grade, Electron Microscopy Sciences) in phosphate buffer (pH 7.4) for 4 h, then post-fixed with 1% osmium tetroxide (Electron Microscopy Sciences), dehydrated to 100% ethanol and critical-point dried with liquid CO2. Gold-palladium-coated specimens were examined with a Jeol JSM-6301FE microscope operated at 5 kV [37].
Acridine orange and immunofluorescence staining of larval eye discs
Acridine orange staining of third-instar larval eye discs was performed as previously described [38]. Quantification of acridine orange-positive cell was performed by Image-Pro Plus 5.1 (Media Cybernetics). Immunofluorescence staining was performed as previously described [16]. Rabbit anti-SARS-virus-PUPM antibody (C-term) (1:100; Abgent) and goat anti-rabbit IgG (H+L)–FITC secondary antibody (1:250; Zymed) were used. Propidium iodide (10 μg/ml, Molecular Probes) was used to label cell nuclei. Images were captured using an Olympus BX51 upright fluorescence microscope or a Leica NT confocal microscope.
Western blot analysis
For fly protein sample preparation, 16 adult fly heads of appropriate genotypes were homogenized in 75 μl of 6× SDS sample buffer. SDS–PAGE separation and Western blotting were performed as previously described [16]. Primary antibodies used include phospho-Drosophila Akt (Ser505) antibody (1:1000; Cell Signaling), total Akt antibody (1:1000; Cell Signaling) and anti-β-tubulin E7 (1:2000; Developmental Studies Hybridoma Bank, under the auspices of the NICHD and maintained by The University of Iowa, Department of Biological Sciences, Iowa City, IA 52242, USA). Secondary antibodies used were goat anti-rabbit IgG-HRP antibody (1:2000; Cell Signaling) and goat anti-mouse IgG-HRP antibody (1:2000; Zymed).
Statistical analyses
Statistical analyses were performed using Student’s t test. Data were presented as means+SEM. p values < 0.05 were considered statistically significant.
Results
Expression and subcellular localization of the SARS-CoV Membrane protein in HEK293T cells
To study the function of the SARS-CoV Membrane protein, the M ORF of the CUHK-Su10 SARS-CoV isolate was subcloned into a mammalian expression vector pcDNA3.1(+). By RT-PCR, mRNA expression of the M gene was detected 48 h post-transfection (Fig. 1 A). Immunofluorescence was performed to determine the subcellular localization of the M protein. We observed that M protein displayed a punctate localization in the cytoplasm (Fig. 1E) and was partially co-localized with the Golgi body (Fig. 1G, inset).
Fig. 1.

Expression and subcellular localization of SARS-CoV Membrane protein in HEK293T cells. (A) mRNA expression level of the SARS-CoV M gene was examined by RT-PCR. Cells transfected with pcDNA3.1(+)-Membrane plasmid showed M mRNA expression at 48 h post-transfection, while no expression was detected in the untransfected and empty vector controls. GAPDH was used as loading control. RT(+) and RT(−) represent the presence and absence of reverse transcriptase enzyme in the RT reaction, respectively. (B–G) Subcellular localization of M protein was determined by immunofluorescence staining. The M protein displayed a punctate cytoplasmic staining pattern (E, in green) and partially localized (G) with the Golgi body (F, in red). Untransfected cells showed no expression of Membrane protein (B). Scale bars represent 16 μm.
Induction of apoptosis by SARS-CoV Membrane protein in HEK293T cells
When the cell nuclei of M-transfected HEK293 cells were stained with Hoechst dye, we found that ∼56% of the cell nuclei were condensed as indicated by size reduction (Fig. 2 I and M) whereas only around 10% of the control cells had condensed nuclei (Fig. 2B, E and M). Since nuclear condensation is a hallmark feature of apoptosis, we reasoned that M over-expression induced apoptosis in HEK293T cells. To confirm this, we treated untransfected HEK293T cells with staurosporine, a commonly used apoptosis inducer, and nuclear condensation was also observed (Fig. 2K and M).
Fig. 2.

Induction of apoptosis by over-expression of M protein in HEK293T cells. (A–L) M protein expression induced nuclear condensation in HEK293T cells. Expression of M protein was only detected in transfected cells (G, in green), but not in untransfected (A and J) and empty vector (D) controls. Hoechst 33342 was used to label cell nuclei (B, E, H and K, in blue). Cells expressed with M protein showed nuclear condensation (I, arrowheads) at 48 h post-transfection. Untransfected cells treated with 1 μM staurosporine for 8 h also displayed nuclear condensation (K, arrowheads), while untreated (B) and empty vector (E) controls showed normal nuclear morphology. (M) The percentage of cells showed nuclear condensation was quantified. Results were plotted as percentage of cells showed nuclear condensation and expressed as means + SEM of three independent experiments. At least 100 cells were counted in each experiment. Scale bars represent 16 μm. ∗p < 0.05. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this paper.)
Since cytochrome c release from the mitochondria represents one form of apoptotic cell death, we performed cytochrome c immunofluorescence staining to examine whether mitochondrial release of cytochrome c was detected in M-transfected cells. Unlike the untransfected control (Fig. 3 A), we found that cells either transfected with M (Fig. 3C) or treated with staurosporine (Fig. 3E) showed extensive mis-localization of cytochrome c protein which is indicative of mitochondrial release of cytochrome c protein [39]. The appearance of M-transfected (Fig. 3D) and staurosporine-treated cells (Fig. 3F) were also morphologically distinct from the untransfected control (Fig. 3B). We further showed that M-induced nuclear condensation in HEK293T cells could be suppressed by caspase-3, -8 and -9 inhibitors (Fig. 3G).
Fig. 3.

Cytochrome c and caspases are involved in M-induced apoptosis. (A–F) M protein expression induced mis-localization of cytochrome c protein in HEK293T cells. When cells were either transfected with M (C) for 48 h or treated with 1 μM staurosporine for 8 h (E), mis-localization of cytochrome c protein was observed. Untransfected cells were used as control (A). Respective phase-contrast images are shown in B, D and F. Scale bars represent 16 μm. (G) M-induced nuclear condensation was inhibited by caspase inhibitors (at 50 μM) z-DQMD-fmk (caspase-3 inhibitor V), z-IETD-fmk (caspase-8 inhibitor II) and z-LEHD-fmk (caspase-9 inhibitor I). Results were plotted as percentage of cells showed nuclear condensation and expressed as means + SEM of three independent experiments. At least 100 cells were counted in each experiment. ∗p < 0.05.
Over-expression of SARS-CoV Membrane protein in Drosophila
We previously used a transgenic Drosophila model [16] to demonstrate the pro-apoptotic role of the SARS-CoV 3a protein [17]. In this study, we established UAS-M transgenic fly lines, and over-expressed the SARS-CoV M protein in the Drosophila eye using GAL4/UAS transgene expression system [36]. Over-expression of M caused a rough-eye phenotype in adult flies, as shown by the loss of regularity of external eye morphology (Fig. 4 C), while the over-expression of the EGFP control protein (Fig. 4B) did not show any obvious dominant eye malformation phenotype. To determine the subcellular localization of M in Drosophila, we performed immunofluorescence staining in third instar larval eye discs (Fig. 4D and F). Similar to mammalian cells (Fig. 1E), M displayed a punctate cytoplasmic localization in fly cells (Fig. 4F), whereas the EGFP control protein showed homogeneous intracellular localization (Fig. 4E).
Fig. 4.
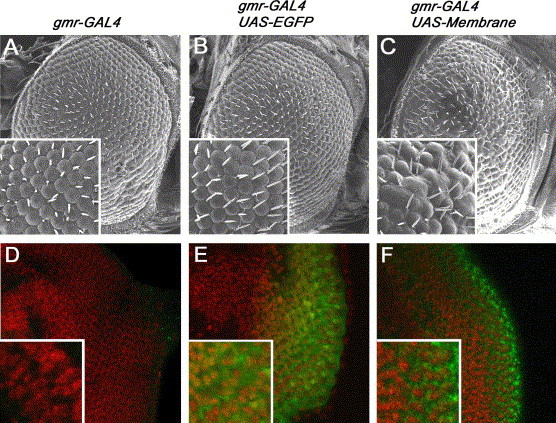
Induction of rough-eye phenotype by over-expression of M protein in Drosophila. (A–C) Over-expression of the M protein in eye tissues resulted in rough-eye phenotype as characterized by loss of regularity of the adult external eye structure (C), whereas the gmr-GAL4 driver alone control (A) and over-expression of the EGFP protein (B) showed no dominant external eye phenotype. (D–F) Subcellular localization of M protein in the Drosophila third instar imaginal eye disc tissues. The M protein showed a distinct punctate cytoplasmic expression pattern (F, in green), whereas the expression EGFP control protein showed homogeneous intracellular staining (E, in green). Cell nuclei were stained by propidium iodide (in red). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this paper.)
Over-expression of SARS-CoV Membrane protein induced apoptosis in Drosophila
Since over-expression of many genes would result in rough-eye phenotype in Drosophila, we therefore performed standard acridine orange (AO) staining [38] in third instar larval imaginal eye discs to examine the pro-apoptotic roles of M in vivo. Acridine orange is a dye that specifically stains apoptotic cells, and we found that the number of AO-positive cells in M-over-expressing eye discs (Fig. 5 G and K) was significantly higher than that of the control (Fig. 5F and K). To further confirm the pro-apoptotic role of M, we co-expressed two anti-apoptotic factors, P35 (a viral caspase inhibitor) and the Drosophila inhibitor of apoptosis 1 (DIAP1) protein, independently with M. As expected, both the rough-eye phenotype (Fig. 5B–D) and elevated number of AO-positive cells (Fig. 5G–I and K) were mostly suppressed back to the control level, respectively.
Fig. 5.

Induction of apoptosis by over-expression of M protein in Drosophila. (A–E) gmr-GAL4 alone control showed no dominant eye phenotype (A). A rough-eye phenotype was observed upon M protein over-expression (B), and was suppressed by co-expression with anti-apoptotic genes DIAP1 (C), P35 (D), and apo-cytochrome cdc3EP2305 (E). (F–J) M over-expression induced apoptosis in third instar imaginal eye discs as indicated by the increase in the number of acridine orange (AO)-stained cells (G). Induction of apoptosis by M protein was suppressed by the co-expression of anti-apoptotic genes DIAP1 (H), P35 (I), and apo-cytochrome cdc3EP2305 (J). Arrows indicate morphogenetic furrows. (K) The number of AO-positive cells in each genotype was quantified. Results were plotted as the number of AO-positive cells and expressed as means + SEM of three-independent experiments. At least nine imaginal eye discs were analyzed in each genotype. ∗p < 0.05 versus gmr-GAL4; #p < 0.05 versus gmr-GAL4 UAS-Membrane.
We next questioned whether M-induced apoptosis would act through the route of cytochrome c in flies as in HEK293T cells (Fig. 3C). It has been reported that over-expression of the apo-form of cytochrome c (without heme group) can block apoptosis [40]. We made use of an EP insert line dc3 EP2305 to over-express the endogenous apo-cytochrome c gene dc3 [16] and asked if dc3 over-expression could suppress M-induced apoptosis in Drosophila. Both the rough-eye phenotype (Fig. 5B) and elevated number of AO-positive apoptotic cells in eye discs (Fig. 5G and K) were largely suppressed upon co-expression of M with apo-cytochrome c (Fig. 5 E, J and K).
Expression of phosphoinositide-dependent kinase-1 dominantly suppressed SARS-CoV Membrane-induced apoptosis
We performed a forward genetic modifier screen in Drosophila in order to gain further insights into the mechanistic details of M action in vivo. By analyzing the modifying effects of overlapping genomic deletion lines, from both the Exelixis [41] and DrosDel [42] collections, on the M-induced rough-eye phenotype (Fig. 4C); 30 modifying genomic regions were identified (C.M. Chan, W.M. Chan, C.S. Chan, C.W. Ma, K.W. Ching, L.L. Ho, K.M. Lau, H.Y. Chan, unpublished observations). Detailed characterization of one of these modifying regions (61B1-C1) is described here. The 61B1-C1 genomic region is uncovered by a deletion line Df(3L)ED201 [42]. We further found that one of the EP insert lines in the 61B1-C1 region, PDK-1 EP837, dominantly suppressed the M-induced rough-eye phenotype (Fig. 6 B and C). The PDK-1 EP837 insert is located at the 5′ upstream region of the phosphoinositide-dependent kinase-1 (PDK-1) gene transcription unit, and had previously been shown to cause over-expression of endogenous PDK-1 [35].
Fig. 6.

Suppression of M-induced apoptosis by phosphoinositide-dependent kinase-1 over-expression in Drosophila. (A–C) PDK-1 over-expression suppressed M-induced rough-eye phenotype. gmr-GAL4 alone control showed no dominant eye phenotype (A). The M-induced rough-eye phenotype (B) was suppressed by over-expression of PDK-1 (C). In the presence of GAL4, PDK-1EP837 causes over-expression of endogenous PDK-1[35]. (D–F) PDK-1 over-expression suppressed M-induced apoptosis as shown by acridine orange (AO) staining. The elevated number of AO-positive cells caused by M protein over-expression in imaginal eye discs (E) was reduced upon the over-expression of PDK-1 (F). Arrows indicate morphogenetic furrows. (G) The number of AO-positive cells in each genotype was quantified. Results were plotted as the number of AO-positive cells and expressed as means + SEM of three-independent experiments. At least nine imaginal eye discs were analyzed in each genotype. ∗p < 0.05 versus gmr-GAL4; #p < 0.05 versus gmr-GAL4 UAS-Membrane.
Since PDK-1 over-expression was able to rescue M-induced rough-eye phenotype, we investigated if PDK-1 suppressed the phenotype through inhibiting apoptosis. We showed that over-expression of SARS-CoV M induced apoptosis in vivo as indicated by the increased number of AO-positive cells (Figs. 5G and 6E). When M was co-expressed with PDK-1, the number of AO-positive cells was largely reduced (Fig. 6F and G).
Down-regulation of Akt phosphorylation by SARS-CoV Membrane protein in vivo
Both Akt and PDK-1 kinases play a central role in the cell survival pathway [43]. Since the Akt pathway can modulate apoptosis [44], we therefore investigated whether over-expression of Drosophila Akt1 could suppress M-induced apoptosis. Unlike PDK-1, the M-induced rough-eye phenotype was not suppressed by Akt1 over-expression (data not shown). Since the phosphorylation state of Akt is an essential cell survival indicator, we then argued if M-over-expression would affect Akt1 phosphorylation. Using phospho-specific Akt1 (Ser505) antibodies, we found that over-expression of M down-regulated the phosphorylation states of Akt1 (Fig. 7 A and B) without affecting total Akt1 protein levels (Fig. 7A and C).
Fig. 7.

Down-regulation of Akt protein phosphorylation in transgenic Drosophila over-expressing M protein. (A) Western blot analysis showed that M expression reduced Akt protein phosphorylation without altering total Akt protein level. β-tubulin was used as loading control. Phospho- (B) and total-Akt protein (C) levels were quantified and normalized against the β-tubulin loading control. Results were plotted as the percentage change of band intensity and expressed as means + SEM of five independent experiments. ∗p < 0.05.
Discussion
The SARS-CoV Membrane protein is the most abundant protein embedded in the viral envelope [25]. The M protein promotes membrane fusion, regulates viral replication, and is also responsible for the packing of viral RNA into matured virions [26], [33]. It has been shown that M is concentrated in the Golgi body of viral-infected cells [45]. Time-lapse microscopy further shows that the M protein is transported in and out of the Golgi body via trafficking vesicles in mammalian cells [46], which further demonstrates its dynamic trafficking properties. In the present study, we showed that M protein partially localized with the Golgi body (Fig. 1G) which is consistent with previous findings [46], [47]. The incomplete co-localization of M with the Golgi body may be explained by the dynamic shuttling properties of the M protein as previously observed [46].
We utilized both in vitro (HEK293T) and in vivo (transgenic Drosophila) models to delineate the functions of the SARS-CoV M protein. HEK293T cells are commonly used in the functional studies of SARS-CoV proteins [21], [30], [48], [49], [50], [51], [52]. Because of its powerful genetics, Drosophila has been used as an in vivo model to study viral gene functions [53], [54], [55], [56]. Previously, our group established a transgenic Drosophila model to study the roles of the SARS-CoV 3a gene product [16]. We showed that over-expression of 3a induced apoptosis in Drosophila, and the results were consistent with a parallel investigation in mammalian cells [17]. In addition, co-expression of 3a, M and E proteins in insect cells can promote the formation of viral-like particles [57]. All these findings illustrate that non-mammalian systems, particularly Drosophila in this study, are plausible models for the studies of viral gene functions in vivo.
It is well known that SARS-CoV infection induces apoptosis in viral-infected cells [4], [11], [13], [14], [15] and the pro-apoptotic properties of a number of individual viral proteins have recently been revealed [12], [16], [17], [18], [19], [20], [21], [22]. Common morphological apoptotic features including cell shrinkage, condensed chromatin and apoptotic body formation are all observed in SARS-CoV viral-infected cells [13]. Swelling of mitochondria is also found in viral-infected cells [13] which indicates a mitochondrial involvement in SARS-CoV-induced apoptosis. At the mechanistic level, SARS-CoV infection has been shown to interfere with the Bcl-2 anti-apoptotic pathway [15]. In this study, we observed that both the in vitro (Fig. 2) and in vivo (Fig. 5) over-expression of M protein induced apoptotic cell death. We also observed nuclear condensation (Fig. 2H) and mis-localization of mitochondrial cytochrome c protein in M-expressing cells (Fig. 3C). Our findings are therefore well in line with the previously described pro-apoptotic properties of SARS-CoV.
In apoptotic cells, holo-cytochrome c protein released from mitochondria triggers the formation of apoptosome and activates the caspase cascades to promote apoptosis [58]. It was reported that the expression of apo-cytochrome c protein (without heme group) is able to compete with holo-cytochrome c (with heme group) for Apaf1, which in turn blocks caspase activation and inhibits apoptosis [40]. We showed that over-expression of apo-cytochrome c blocked M-induced apoptosis (Fig. 5E, J and K, [16]). Caspase activation has been shown to be involved in SARS-CoV-induced apoptosis [15]. Previous studies also revealed that SARS-CoV viral proteins are able to activate individual caspases. For example, N activates caspase-3 and -7 [12], 3a activates caspase-8 [17], and 7a activates caspase-3 [18]. To test the involvement of caspases in M-induced apoptosis, we treated M-expressing cells with caspase inhibitors and found that M-induced nuclear condensation could be prevented by inhibitors specific for caspase-3, -8 and -9 (Fig. 3G). Drosophila inhibitor of apoptosis 1 (DIAP1) and the baculoviral caspase inhibitor protein P35 are effective broad-range inhibitors of caspase activation in Drosophila [59], [60]. When we co-expressed M with either DIAP1 (Fig. 5H and K) or P35 (Fig. 5I and K) in vivo, we found that M-induced apoptosis was suppressed. The incomplete inhibition of apoptosis by specific caspases-3, -8 and -9 inhibitors (Fig. 3G) underscores the involvement of other caspases in M-induced apoptosis.
The Akt pathway is a cellular pro-survival signaling cascade [61], and is known to regulate mitochondria-mediated apoptosis [62], [63], [64], [65]. Caspase-9 is one of the phosphorylation targets of Akt, and the protease activity of caspase-9 is reduced upon Akt phosphorylation [66]. In addition, it is known that caspase-8 activity is also regulated indirectly through Akt [67]. The Akt pathway has been implicated in viral infections [24] including SARS-CoV [5], [9], [11]. A weak activation of the Akt cellular pro-survival pathway has been observed in viral-infected cells as evidenced by transient phosphorylation of Akt, GSK-3β and PKCζ kinases [5], [9], [11]. Such transient activation would reflect the inability of viral-infected cells to continuously combat with viral infection. In this report, we showed that over-expression of the M protein caused down-regulation of Akt phosphorylation without affecting total Akt protein level (Fig. 7). Such action would therefore contribute to the reduction of the cell survival signal, which in turn lead to the apoptotic induction.
From our forward genetic modifier screen, we identified PDK-1 kinase, an upstream effector in the Akt signaling pathway, as a dominant suppressor of M-induced rough-eye phenotype (Fig. 6C) and apoptosis (Fig. 6F). In addition, we found that heterozygous loss-of-function mutation of PDK-1 (PDK-1 1, PDK-1 2 [35]) showed no modification on the M-induced rough-eye phenotype (data not shown). This indicates that PDK-1 showed no haplo-insufficient effect on M function. The human homolog of PDK-1 gene is expressed in the lung which further implicates its role in SARS-CoV infection (http://www.ncbi.nlm.nih.gov/UniGene/ESTProfileViewer.cgi?uglist=Hs.459691) [68].
PDK-1 is responsible for the phosphorylation of Thr but not Ser residue of Akt [69]. Since only Ser phosphorylation of Akt is found to be altered by SARS-CoV [5], [9], [11], this suggests that PDK-1 activity on Akt phosphorylation is probably not a major target of SARS-CoV.
It is now known that activation of apoptosis induces CPE in SARS-CoV-infected cells [13]. The M protein has previously been shown to interact with a number of SARS-CoV proteins, including N [27], [28], [29], S [30], 3a [31] and 7a [32] to carry out a variety of viral pathologic processes. Since all the above mentioned M-interacting viral proteins are pro-apoptotic, it is legitimate to speculate that the SARS-CoV utilizes multiple viral proteins to ensure that apoptosis is executed in infected cells. Understanding the cellular pathways these pro-apoptotic viral proteins modulate in viral-infected cell is essential for the development of comprehensive strategies to mitigate SARS-CoV-induced CPE.
Acknowledgments
We thank Priscilla Chan, Alan Wong and Steve Lo for critical reading of the manuscript; Priscilla Chan, C.S. Chan, Gary Ching, Lilian Ho and K.M. Lau for performing forward genetic modifier screen; and Jongkyeong Chung for fly lines. This work was supported by a CUHK RGC Research Grant Direct Allocation (2030334).
Footnotes
Abbreviations used: AO, acridine orange; CPE, cytopathic effect; DIAP1, Drosophila inhibitor of apoptosis 1; E, Envelope; EGFP, enhanced green fluorescent protein; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GSK3β, glycogen synthase kinase3β; IAP, inhibitor of apoptosis; M, Membrane; MAPK, mitogen-activated protein kinase; N, Nucleocapsid; ORF, opening reading frame; PDK-1, phosphoinositide-dependent kinase-1; PKCζ, protein kinase Cζ; S, Spike; SARS-CoV, Severe Acute Respiratory Syndrome-Coronavirus; Ser, serine; Thr, threonine.
References
- 1.Marra M.A. Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- 2.Rota P.A. Science. 2003;300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 3.Ksiazek T.G. N. Engl. J. Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 4.Mizutani T., Fukushi S., Saijo M., Kurane I., Morikawa S. Biochem. Biophys. Res. Commun. 2004;319:1228–1234. doi: 10.1016/j.bbrc.2004.05.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mizutani T., Fukushi S., Saijo M., Kurane I., Morikawa S. FEBS Lett. 2006;580:1417–1424. doi: 10.1016/j.febslet.2006.01.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee C.H. J. Immunol. 2004;172:7841–7847. doi: 10.4049/jimmunol.172.12.7841. [DOI] [PubMed] [Google Scholar]
- 7.Mizutani T. Biochem. Biophys. Res. Commun. 2006 [Google Scholar]
- 8.Mizutani T., Fukushi S., Murakami M., Hirano T., Saijo M., Kurane I., Morikawa S. FEBS Lett. 2004;577:187–192. doi: 10.1016/j.febslet.2004.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mizutani T., Fukushi S., Saijo M., Kurane I., Morikawa S. Virology. 2004;327:169–174. doi: 10.1016/j.virol.2004.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mizutani T., Fukushi S., Saijo M., Kurane I., Morikawa S. Biochim. Biophys. Acta. 2005;1741:4–10. doi: 10.1016/j.bbadis.2005.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mizutani T. FEMS Immunol. Med. Microbiol. 2006;46:236–243. doi: 10.1111/j.1574-695X.2005.00028.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Surjit M., Liu B., Jameel S., Chow V.T., Lal S.K. Biochem. J. 2004;383:13–18. doi: 10.1042/BJ20040984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yan H., Xiao G., Zhang J., Hu Y., Yuan F., Cole D.K., Zheng C., Gao G.F. J. Med. Virol. 2004;73:323–331. doi: 10.1002/jmv.20094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ren L., Yang R., Guo L., Qu J., Wang J., Hung T. DNA Cell Biol. 2005;24:496–502. doi: 10.1089/dna.2005.24.496. [DOI] [PubMed] [Google Scholar]
- 15.Bordi L. Arch. Virol. 2006;151:369–377. doi: 10.1007/s00705-005-0632-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wong S.L. Biochem. Biophys. Res. Commun. 2005;337:720–729. doi: 10.1016/j.bbrc.2005.09.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Law P.T. J. Gen. Virol. 2005;86:1921–1930. doi: 10.1099/vir.0.80813-0. [DOI] [PubMed] [Google Scholar]
- 18.Tan Y.J., Fielding B.C., Goh P.Y., Shen S., Tan T.H., Lim S.G., Hong W. J. Virol. 2004;78:14043–14047. doi: 10.1128/JVI.78.24.14043-14047.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin C.W., Lin K.H., Hsieh T.H., Shiu S.Y., Li J.Y. FEMS Immunol. Med. Microbiol. 2006;46:375–380. doi: 10.1111/j.1574-695X.2006.00045.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chow K.Y., Yeung Y.S., Hon C.C., Zeng F., Law K.M., Leung F.C. FEBS Lett. 2005;579:6699–6704. doi: 10.1016/j.febslet.2005.10.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yuan X., Shan Y., Zhao Z., Chen J., Cong Y. Virol. J. 2005;2:66. doi: 10.1186/1743-422X-2-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang Y. Biochem. J. 2005;392:135–143. doi: 10.1042/BJ20050698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Surjit M., Kumar R., Mishra R.N., Reddy M.K., Chow V.T., Lal S.K. J. Virol. 2005;79:11476–11486. doi: 10.1128/JVI.79.17.11476-11486.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cooray S. J. Gen. Virol. 2004;85:1065–1076. doi: 10.1099/vir.0.19771-0. [DOI] [PubMed] [Google Scholar]
- 25.He Y., Zhou Y., Siddiqui P., Niu J., Jiang S. J. Clin. Microbiol. 2005;43:3718–3726. doi: 10.1128/JCM.43.8.3718-3726.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hu Y. Genomics Proteomics Bioinformatics. 2003;1:118–130. doi: 10.1016/S1672-0229(03)01016-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luo H., Wu D., Shen C., Chen K., Shen X., Jiang H. Int. J. Biochem. Cell Biol. 2006;38:589–599. doi: 10.1016/j.biocel.2005.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fang X., Ye L., Timani K.A., Li S., Zen Y., Zhao M., Zheng H., Wu Z. J. Biochem. Mol. Biol. 2005;38:381–385. doi: 10.5483/bmbrep.2005.38.4.381. [DOI] [PubMed] [Google Scholar]
- 29.He R. Virus Res. 2004;105:121–125. doi: 10.1016/j.virusres.2004.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang Y., Yang Z.Y., Kong W.P., Nabel G.J. J. Virol. 2004;78:12557–12565. doi: 10.1128/JVI.78.22.12557-12565.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tan Y.J. J. Virol. 2004;78:6723–6734. doi: 10.1128/JVI.78.13.6723-6734.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fielding B.C. Biochem. Biophys. Res. Commun. 2006;343:1201–1208. doi: 10.1016/j.bbrc.2006.03.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ho Y., Lin P.H., Liu C.Y., Lee S.P., Chao Y.C. Biochem. Biophys. Res. Commun. 2004;318:833–838. doi: 10.1016/j.bbrc.2004.04.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huen N.Y., Chan H.Y. Biochem. Biophys. Res. Commun. 2005;334:1074–1084. doi: 10.1016/j.bbrc.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 35.Cho K.S. Proc. Natl. Acad. Sci. USA. 2001;98:6144–6149. doi: 10.1073/pnas.101596998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brand A.H., Perrimon N. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- 37.Chau K.W., Chan W.Y., Shaw P.C., Chan H.Y. Biochem. Biophys. Res. Commun. 2006;346:150–159. doi: 10.1016/j.bbrc.2006.05.112. [DOI] [PubMed] [Google Scholar]
- 38.Hay B.A., Wassarman D.A., Rubin G.M. Cell. 1995;83:1253–1262. doi: 10.1016/0092-8674(95)90150-7. [DOI] [PubMed] [Google Scholar]
- 39.Arnoult D., Parone P., Martinou J.C., Antonsson B., Estaquier J., Ameisen J.C. J. Cell Biol. 2002;159:923–929. doi: 10.1083/jcb.200207071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martin A.G., Fearnhead H.O. J. Biol. Chem. 2002;277:50834–50841. doi: 10.1074/jbc.M209369200. [DOI] [PubMed] [Google Scholar]
- 41.Parks A.L. Nat. Genet. 2004;36:288–292. doi: 10.1038/ng1312. [DOI] [PubMed] [Google Scholar]
- 42.Ryder E. Genetics. 2004;167:797–813. doi: 10.1534/genetics.104.026658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Downward J. Curr. Opin. Cell Biol. 1998;10:262–267. doi: 10.1016/s0955-0674(98)80149-x. [DOI] [PubMed] [Google Scholar]
- 44.Beere H.M. J. Clin. Invest. 2005;115:2633–2639. doi: 10.1172/JCI26471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qinfen Z., Jinming C., Xiaojun H., Huanying Z., Jicheng H., Ling F., Kunpeng L., Jingqiang Z. J. Med. Virol. 2004;73:332–337. doi: 10.1002/jmv.20095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nal B. J. Gen. Virol. 2005;86:1423–1434. doi: 10.1099/vir.0.80671-0. [DOI] [PubMed] [Google Scholar]
- 47.Voss D., Kern A., Traggiai E., Eickmann M., Stadler K., Lanzavecchia A., Becker S. FEBS Lett. 2006;580:968–973. doi: 10.1016/j.febslet.2006.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yuan X. Virology. 2006;346:74–85. doi: 10.1016/j.virol.2005.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li W. Nature. 2003;426:450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Simmons G., Reeves J.D., Rennekamp A.J., Amberg S.M., Piefer A.J., Bates P. Proc. Natl. Acad. Sci. USA. 2004;101:4240–4245. doi: 10.1073/pnas.0306446101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Giroglou T., Cinatl J., Jr., Rabenau H., Drosten C., Schwalbe H., Doerr H.W., von Laer D. J. Virol. 2004;78:9007–9015. doi: 10.1128/JVI.78.17.9007-9015.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Qin Z.L., Zhao P., Zhang X.L., Yu J.G., Cao M.M., Zhao L.J., Luan J., Qi Z.T. Biochem. Biophys. Res. Commun. 2004;324:1186–1193. doi: 10.1016/j.bbrc.2004.09.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Adamson A.L., Wright N., LaJeunesse D.R. Genetics. 2005;171:1125–1135. doi: 10.1534/genetics.105.042572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Battaglia P.A., Ponti D., Naim V., Venanzi S., Psaila R., Gigliani F. Cell Motil. Cytoskeleton. 2005;61:129–136. doi: 10.1002/cm.20070. [DOI] [PubMed] [Google Scholar]
- 55.Battaglia P.A., Zito S., Macchini A., Gigliani F. J. Cell Sci. 2001;114:2787–2794. doi: 10.1242/jcs.114.15.2787. [DOI] [PubMed] [Google Scholar]
- 56.Leulier F., Marchal C., Miletich I., Limbourg-Bouchon B., Benarous R., Lemaitre B. EMBO Rep. 2003;4:976–981. doi: 10.1038/sj.embor.embor936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shen S., Lin P.S., Chao Y.C., Zhang A., Yang X., Lim S.G., Hong W., Tan Y.J. Biochem. Biophys. Res. Commun. 2005;330:286–292. doi: 10.1016/j.bbrc.2005.02.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Saelens X., Festjens N., Vande Walle L., van Gurp M., van Loo G., Vandenabeele P. Oncogene. 2004;23:2861–2874. doi: 10.1038/sj.onc.1207523. [DOI] [PubMed] [Google Scholar]
- 59.Stennicke H.R., Ryan C.A., Salvesen G.S. Trends Biochem. Sci. 2002;27:94–101. doi: 10.1016/s0968-0004(01)02045-x. [DOI] [PubMed] [Google Scholar]
- 60.Callus B.A., Vaux D.L. Cell Death Differ. 2007;14:73–78. doi: 10.1038/sj.cdd.4402034. [DOI] [PubMed] [Google Scholar]
- 61.Downward J. Semin. Cell Dev. Biol. 2004;15:177–182. doi: 10.1016/j.semcdb.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 62.Tsuruta F., Masuyama N., Gotoh Y. J. Biol. Chem. 2002;277:14040–14047. doi: 10.1074/jbc.M108975200. [DOI] [PubMed] [Google Scholar]
- 63.Datta S.R., Dudek H., Tao X., Masters S., Fu H., Gotoh Y., Greenberg M.E. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 64.del Peso L., Gonzalez-Garcia M., Page C., Herrera R., Nunez G. Science. 1997;278:687–689. doi: 10.1126/science.278.5338.687. [DOI] [PubMed] [Google Scholar]
- 65.Kennedy S.G., Kandel E.S., Cross T.K., Hay N. Mol. Cell Biol. 1999;19:5800–5810. doi: 10.1128/mcb.19.8.5800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cardone M.H., Roy N., Stennicke H.R., Salvesen G.S., Franke T.F., Stanbridge E., Frisch S., Reed J.C. Science. 1998;282:1318–1321. doi: 10.1126/science.282.5392.1318. [DOI] [PubMed] [Google Scholar]
- 67.Skurk C., Maatz H., Kim H.S., Yang J., Abid M.R., Aird W.C., Walsh K. J. Biol. Chem. 2004;279:1513–1525. doi: 10.1074/jbc.M304736200. [DOI] [PubMed] [Google Scholar]
- 68.Tong Z., Wu X., Chen C.S., Kehrer J.P. Lung. Cancer. 2006;52:117–124. doi: 10.1016/j.lungcan.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 69.Scheid M.P., Woodgett J.R. FEBS Lett. 2003;546:108–112. doi: 10.1016/s0014-5793(03)00562-3. [DOI] [PubMed] [Google Scholar]