

Abstract
Objectives
Parental death during childhood, and offspring and spouse death during adulthood have individually been associated with faster cognitive decline and higher Alzheimer’s disease (AD) risk in late-life. However, the cumulative effect of childhood and adulthood family deaths on AD risk among different age cohorts has not been studied.
Methods
To examine these associations, this prospective cohort study uses a population-based sample of 4,545 initially non-demented participants (56.7% female; age M=75.0/SD=6.9 years) observed at four triennial waves, linked with objective Utah Population Database data on cumulative mother, father, sibling, spouse and offspring death experienced during childhood and adulthood. Cox regression modeled survival time from baseline interview to AD onset, as a function of family deaths during childhood or adulthood, among different age groups, along with gender and presence of ε4 allele at apolipoprotein E (APOE) polymorphic genetic locus.
Results
Age group significantly moderated the relationship between family death and AD; among persons aged 65–69 years at baseline (children of the Great Depression), those exposed to 3–4 deaths and 5+ deaths during adulthood exhibited a doubling of AD risk (adjusted Hazard Ratio, aHR=2.25, p=.038 and aHR=2.72, p=.029), while among persons aged 80 years and older, those exposed to 3–4 deaths during adulthood exhibited lower AD risk (HR=0.539, p=0.014). In a combined model of childhood and adulthood deaths, these findings persisted.
Conclusions
Results suggest a cohort effect in the link between family member deaths during adulthood and AD risk later in life.
Keywords: Alzheimer’s Disease, Psychosocial Stressors, Lifespan Development
INTRODUCTION
Alzheimer’s disease (AD) is a growing public health concern, with a prevalence of 11–16 million cases projected by 2050 in the US alone (Alzheimer’s Association, 2012), and 115.4 million worldwide (World Health Organization, 2012). A mechanism that may increase risk of AD is chronic exposure to psychosocial stressors. Such stressors are associated with poor mental and physical health outcomes, including depression (Tennant, 2002), heart problems (Vitaliano et al., 2002), and diabetes (Lloyd et al., 2005), which in turn are risk factors for AD. Research also indicates a direct link between stress and AD; although psychosocial stress can restore allostasis to the body by releasing catecholamines and glucocorticoids (McEwen, 2002), an accumulation, absence, and/or mismanagement of this reactivity-recovery process leads to neural death in the CA3 region of the hippocampus (Meaney et al., 1988). Although these biological neurodegenerative changes occur over the life course they are often not clinically detectable until decades after their onset (Braak and Braak, 1997).
The effects of exposure to stress may depend on the developmental period of the individual and on sociocultural factors. Animal models have found the effect of early maternal separation on lower hippocampal and glial numbers to persist into later in life (Leventopoulos et al., 2007). However, animal and human studies on resilience also indicate that early-life stress can protect against the adverse effects of later-life stress on hippocampal plasticity (Oomen et al., 2010), and anxiety and cortisol levels (Lyons et al., 2009). Sociocultural factors (“cohort effects”), such as child poverty during the Great Depression (Elder, 1974) may also impact the effect of stressors. For instance, Yang (2007) reported significant cohort and age-by-cohort interactions for trajectories of late life depressive symptoms.
One of the most devastating and life altering stressors is death of a close relative. In addition to affecting well-being (Buckley et al., 2009), this stressor has also been linked to risk for dementia. For instance, studies have found parental death during childhood (Persson and Skoog, 1996) and maternal death during adolescence (Norton et al., 2011) to be associated with six-fold and two-fold increased risk for dementia and AD, respectively. Other large-scale epidemiological studies also demonstrate the link between parental death and AD or dementia (Whalley et al., 2013, Tsolaki et al., 2010). A study by Ravona-Springer and colleagues (2012) found that persons who experienced crisis after parental death at ages 0–6 years, 7–12 years, and 13–18 years exhibited 3.06, 2.15, and 2.35 greater odds of dementia, respectively, suggesting the effect of parental death to be augmented at younger age. Offspring death can also be devastating and life altering, and can result in bereavement lasting many years (Li et al., 2003) and faster rate of late-life cognitive decline (Greene et al., 2014). Another major loss, widowhood, has also been linked with dementia risk (Fratiglioni et al., 2000; Hatch et al., 2015). For instance, a longitudinal study of elderly men found that those who had become widowed within the preceding five years showed double the rate of cognitive decline compared to men who stayed married (van Gelder et al., 2006), and Fratiglioni et al. (2000) found that older people who were single or living alone (including divorced and widowed persons) had double the risk for dementia, compared to married persons.
Overall, this literature indicates strong associations between individual familial deaths and risk for dementia and AD. However, no study has examined the cumulative effect of all familial deaths, including death of parents, sibling(s), spouse(s) and offspring, and no examination of such effects during different developmental periods- childhood vs. adulthood, and in different age cohorts. We report herein secondary analysis of a population-based prospective cohort study of dementia with 5,000+ participants where objective data sources were used to ascertain familial deaths. We hypothesized that greater exposure to familial deaths would be associated with greater AD risk.
METHODS
Study Design
The Cache County Memory Study (CCMS) is a population-based prospective cohort study of dementia and its genetic and environmental risk factors. In contrast with case-control studies, which identify cases and matched controls at the time of disease (Song and Chung, 2010), this study identifies a cohort of potential cases and controls prospectively before the time of disease in a population of persons of older age, a known risk factor for AD and other dementias. This cohort consisted of 5,092 participants from the population of 5,654 eligible permanent residents of Cache County, Utah aged 65 years and older on January 1, 1995 who were identified from a sampling frame of Medicare enrollee lists provided by the Health Care Financing Administration. This 90% response rate greatly reduces bias due to non-responders, who tend to be less educated and to have greater cognitive impairment (Norton et al., 1994), known risk factors for AD and other dementias. Four triennial waves of dementia ascertainment were completed in 1995–7, 1998–2000, 2002–4, and 2005–7. To assess the effect of family-related life stressors, CCMS data was linked with retrospective data from the Utah Population Database (UPDB), one of the world’s foremost genealogical databases. The current study examined the association between family deaths during childhood and adulthood and risk for development of incident Alzheimer’s disease (AD) at any of the three follow-up assessments.
Subjects
Beginning with 5,092 participants in CCMS, we removed 359 with prevalent dementia and another 188 with incomplete dementia ascertainment, for a final sample size of 4,545. Of these, 396 (8.7%) developed incident AD, while 187 (4.1%) developed incident non-AD dementia and 3,962 (87.2%) were never diagnosed with dementia at any stage of dementia screening. These latter two groups served as the reference category.
Alzheimer’s Disease Diagnostic Procedures
The CCMS utilized an extensive multistage dementia ascertainment protocol described elsewhere (Breitner et al., 1999), repeated in four triennial waves. At the initial stage of assessment, subjects were administered the Modified Mini-Mental State examination (3MS; Tschanz et al., 2002) and also for those whose 3MS score was below 60 out of 100, the Informant Questionnaire for Cognitive Decline in the Elderly (Jorm and Jacomb, 1989). Participants who screened positive for possible dementia completed an in-depth clinical assessment (CA), along with a 19% sub-sample of “designated controls.” Trained nurses and psychometric technicians administered the CA, which included a one-hour battery of neuropsychological tests, a detailed history of medical and cognitive symptoms, and a structured neurological examination.
A geriatric psychiatrist and neuropsychologist reviewed these data, upon which subjects with a working diagnosis of dementia (per DSM-III-R; American Psychiatric Association, 1987) were selected for psychiatrist examinations and laboratory studies including neuroimaging, and an 18-month follow-up CA. A consensus panel of experts in neurology, geriatric psychiatry, neuropsychology, and cognitive neuroscience reviewed all available data and assigned final consensus diagnoses. Alzheimer’s Disease (AD) was diagnosed according to NINCDS-ADRDA criteria (McKhann et al., 1984). Of the 396 diagnosed cases of incident AD, there were 335 “pure AD” cases, another 44 AD cases that were comorbid with vascular dementia (VaD; per NINDS-AIREN criteria; Roman et al., 1993), and another 17 AD cases that were comorbid with other dementia. Dementia onset was defined as the year in which a participant unambiguously met DSM-III-R criteria for dementia. The reference category was no dementia (n=3,962), either after CA or negative screening, or non-AD dementia.
Informed consent was obtained for each interview. All procedures were approved by the Institutional Review Boards (IRB) of Utah State University (USU), Duke University, and Johns Hopkins University. The Utah Population Database (UPDB) usage was approved by the IRB of USU and the University of Utah, and the Utah Resource for Genetic and Epidemiologic Research.
Familial Death Measurement
The UPDB is one of the world’s richest sources of linked population-based information for demographic, genetic, epidemiological, and public health studies. It provides access to over 14 million records or documents representing over 7 million individuals (for more details see http://www.hci.utah.edu/groups/ppr). The UPDB began as a large set of multigenerational Utah family histories, to which genealogy records, cancer records, birth and death certificates, driver license records, and census records have been linked. All but one of the 5,092 participants in the Cache County Memory Study (CCMS) have been linked to the UPDB, permitting the derivation of psychosocial stressor measures, without the potential problem of recall bias common in retrospective self-reports of risk factors (Uher and McGuffin, 2010).
Death dates in the UPDB were used to objectively ascertain a cumulative total of family deaths including death(s) of mother, father, sibling(s), spouse(s), offspring and stillborn offspring, experienced during childhood (birth to age 17.9) and during adulthood (age 18 to study entry). Number of deaths during childhood was coded into exposure groups of 0 vs. 1 vs. 2 or more, with 0 deaths as the reference category. Number of deaths during adulthood was coded into tertiles of 0–2 deaths vs. 3–4 deaths vs. 5 or more, with 0–2 deaths as the reference category. Although the effect of family death may depend on one’s potential exposure to it, this was not characterized in terms of number of family members at time of death, given that the number of family members would change with each death and thus preclude analyzing number of family death as a cumulative total. Rather, potential exposure was characterized in terms of exposure time differences between childhood and adulthood, and of exposure time differences within adulthood (individuals varied from 65 to 105 years of age at baseline), by computing a “family member death rate” (FMDR), in which the grand total number of deaths across the lifespan was divided by the participant’s age at study entry, multiplied by 10 (to yield “deaths per decade”).
Covariates
Apolipoprotein E (APOE) genotype was determined from buccal DNA using polymerase chain reaction (PCR) amplification and a restriction isotyping method described by Saunders and colleagues (Saunders, 1993). APOE genotype was coded as 0 vs. 1 or more ε4 alleles. Education was scored as highest level of education completed in years. Age in years at study entry was coded into 65–69 years, 70–74 years, 75–79 years and 80 years and older, for conceptual categories of young, early-middle, late-middle and late old age, corresponding also to quartiles of the baseline age distribution. Gender was coded 1=male, 2=female. Socioeconomic status was based on parental occupations listed on the birth certificates of participants’ offspring, using a methodology developed by Nam & Powers (NP-SES; 1983).
Data Analysis
Cox regression analysis was conducted to model the association between family member deaths and AD-free survival time from baseline interview to dementia onset or right-censoring. Non-AD dementia cases were retained until the time of diagnosis, at which time they were right-censored. Moving from the simpler to the more complex to enhance understanding, separate models were computed to examine effect of childhood and adulthood family deaths, followed by a joint model that examined these exposures together. An additional model was computed to examine the effect of FMDR. The proportional hazards assumption was tested for each family death exposure variable separately, by testing the interaction between the exposure variable and time, with a non-significant interaction term indicating that the assumption was met. Models included gender, APOE, age group, education and NP-SES as covariates in initial models, but the latter two were removed in all subsequent models as they were non-significant (education p=0.882, NP-SES p=0.461). Moderating effects were tested for age group, and when an interaction effect was significant at p<0.10, stratified models were computed to further examine differential effects by age group (birth cohort). Adjusted Hazard Ratios (aHR) are reported, to reflect adjustment for covariates. SPSS version 21 was used for all analyses.
RESULTS
Baseline age ranged from 65 to 102 years (M=75.0, SD=6.9), with average education and SES of 13.2 (SD=2.9) years and 59.7 (SD=22.6) (Table 1). Subjects were 99% Caucasian with 2,575 (56.7%) females, and 1,356 (29.8%) with at least one copy and 3,098 (68.2%) with zero copies of e4 allele at APOE, with 91 (2.0%) lacking genotype. Cumulative family deaths during childhood included 2,996 (65.9%) with zero deaths, 1,118 (24.6%) with one death and 431 (9.5%) with two or more deaths. In adulthood, there were 1,454 (32.0%) with two or fewer deaths, 1,692 (37.2%) with three or four deaths, and 1,399 (30.8%) with five or more deaths. FMDR ranged from 0 to 2.0 deaths per decade with quartiles of 0.33, 0.51 and 0.72. The number of subjects who developed AD, by age group was: n=43, n=78, n=111, and n=164 for participants aged 65–69, 70–74, 75–79, and 80+, respectively. Proportional hazards assumption was met for both childhood and adulthood exposure variables as evidence by the interaction with time being non-significant (p=0.314 and p=0.967, respectively).
Table 1.
Demographic Characteristics of Participants
| Variable | |
|---|---|
| Baseline age, M(SD) | 75.0 (6.9) |
| Education, M(SD) | 13.2 (2.9) |
| SES, M(SD) | 59.7 (22.6) |
| Gender, n(%) female | 2,575 (56.7%) |
| APOE e4 | |
| missing, n(%) | 91 (2.0%) |
| 0 e4 alleles, n(%) | 3,098 (68.2%) |
| 1+ e4 alleles, n(%) | 1,356 (29.8%) |
| Childhood deaths | |
| 0 deaths, n(%) | 2,996 (65.9%) |
| 1 death, n(%) | 1,118 (24.6%) |
| 2+ deaths, n(%) | 431 (9.5%) |
| Adult deaths | |
| 0–2 deaths, n(%) | 1,454 (32.0%) |
| 3–4 deaths, n(%) | 1,692 (37.2%) |
| 5+ deaths, n(%) | 1,399 (30.8%) |
| AD | |
| No dementia, n(%) | 3,962 (87.2%) |
| Non-AD dementia, n(%) | 187 (4.1%) |
| AD, n(%) | 396 (8.7%) |
Family Deaths during Childhood and Adulthood
In the initial Cox model, there was a trend for moderation of childhood deaths by age group (p=0.092). When separate models were run for each of four age groups, a trend was found for the association between childhood deaths and AD risk for persons aged 65–69 years (Wald=5.79, df=2, p=0.055), whereas no trend was found among persons aged 70–74 years (Wald=2.02, df=2, p=0.364), 75–79 years (Wald=0.63, df=2, p=0.729), and 80 years or older (Wald=4.46, df=2, p=0.108).
In contrast, baseline age group significantly moderated the association between adulthood deaths and AD risk (Wald=13.18, df=6, p=0.040). Among persons aged 65–69 years, associations between adulthood death and AD were found (Wald=6.55, df=2, p=0.038), wherein both exposure to three to four deaths (Wald=5.14, df=1, aHR=2.246, p=0.023) and five or more deaths (Wald=4.77, df=1, aHR=2.718, p=0.029) were associated with more than double AD risk (Figure 1), compared to persons experiencing zero to two deaths. Individuals in the middle two age groups had no significant association between adulthood deaths and AD risk (Wald=1.51, df=2, p=0.470 for persons aged 70–74 years, and Wald=1.21, df=2, p=0.547 for persons aged 75–79 years). Among persons aged 80 years and older, an association was found (Wald=6.19, df=2, p=0.045), wherein lower AD risk occurred among those exposed to three to four deaths (Wald=6.02, df=1, aHR=0.539, p=0.014), while no effect occurred among those exposed to five or more deaths (Wald=1.66, df=1, aHR=0.759, p=0.197; Figure 2).
Figure 1.

Survival Function by Number of Deaths during Adulthood, among 65–69 Year Olds
Figure 2.

Survival Function by Number of Deaths during Adulthood, among 80+ Year Olds
A combined model including both cumulative deaths during childhood and cumulative deaths during adulthood was computed for each age group. In the youngest age group (65–69 years), number of childhood deaths was not significant (Wald=5.33, df=2, p=.070). However, number of adulthood deaths was significant in this age group (Wald=6.31, df=2, p=0.043), in that having three to four (Wald=4.75, df=1, aHR=2.184, p=0.029) or five or more (Wald=4.82, df=1, aHR=2.734, p=0.028) adulthood deaths were associated with a doubling in AD risk. No effects were found for deaths in the second age group (childhood: Wald=1.90, df=2, p=0.386, adulthood: Wald=1.40, df=2, p=0.497) or in the third age group (childhood: Wald=0.83, df=2, p=0.659, adulthood: Wald=1.43, df=2, p=0.489). Among persons aged 80 and older, there was a non-significant trend for childhood deaths (Wald=4.61, df=2, p=0.100), but a significant effect for adulthood deaths (Wald=6.50, df=2, p=0.039), in that three to four adulthood deaths was associated with significantly lower AD risk (Wald=6.48, df=1, aHR=0.525, p=0.011, five or more deaths not significant: Wald=2.40, df=1, p=0.121).
Next, we conducted models testing the interaction between childhood and adulthood deaths in each age group. We found no evidence of an interaction for all four age groups: persons aged 65–69 years (Wald=1.10, df=4, p=0.895), 70–74 years (Wald=0.33, df=4, p=0.988), 75–79 years (Wald=4.41, df=4, p=0.353), or 80 and older (Wald=8.02, df=4, p=0.091).
Family Member Death Rate
Finally, separate Cox models were computed for each age group to examine the association between FMDR and AD risk. Greater FMDR was significantly associated with higher AD risk for persons aged 65–69 years (Wald=7.75, df=1, aHR=5.0, p=0.005), but was unrelated to AD risk for other age groups (70 to 74 years: Wald=3.27, df=1, aHR=0.39, p=0.071; 75 to 79 years: Wald=0.170, df=1, aHR=0.869, p=0.681; 80 years and older: Wald=0.008, df=1, aHR=1.023, p=0.928). To facilitate visual display in a survival curve of FMDR among persons aged 65–69 years, FMDR was categorized into quartiles (see Figure 3). Models using this variable indicated that persons in the third (Wald=5.008, df=1, aHR=2.547, p=0.025) and fourth quartiles (Wald=7.819, df=1, aHR=3.622, p=0.005) had significantly higher AD risk than those in the first quartile (second quartile not significant: Wald=0.091, df=1, aHR=1.158, p=0.762).
Figure 3.
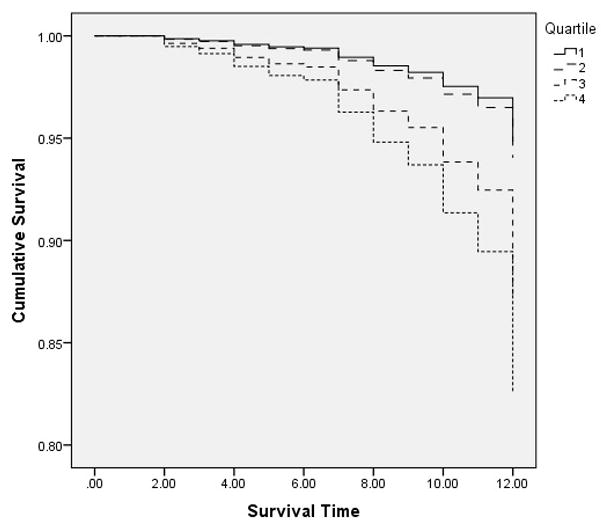
Survival Function by Quartile of Family Member Deaths Per Decade, among 65–69 Year Olds
DISCUSSION
Using a unique population-based epidemiological sample pooled with lifespan historical records, we examined the effects of family member deaths in childhood and adulthood on subsequent AD risk. Although family member deaths during childhood were not linked to AD risk, family deaths during adulthood were associated with increased AD risk. This pattern contrasts with previous findings, which found childhood to be a more vulnerable period than adulthood. This might be partially explained by adulthood spanning a longer exposure time (and hence, accrual of more events/deaths) than childhood. To adjust for this exposure time difference, and for exposure time differences within adulthood, we examined death rate per decade (FMDR). This revealed that individuals with higher exposure (i.e. top two quartiles of FMDR) had heightened AD risk, compared to lower levels of death rate exposure.
Past research has described key mechanisms by which stress is associated with risk for AD, for example, heightened levels of glucocorticoid exposure with long-lasting effects that gradually increase neuronal cell death (Meaney et al., 1988). Our hypothesis that family member deaths over the life course represent major psychosocial stressors and thereby are associated with heightened vulnerability to AD is supported by the current study. Additional investigation can strengthen this hypothesis by identifying the mechanisms by which major life events stressors relate to dementia risk.
We consistently found a birth cohort effect, in that persons aged 65–69 years at study entry formed the only group with strong adverse effects, more than doubling AD risk if exposed to multiple deaths during adulthood. This was also the only age group significantly affected by FMDR. Historical context may be a relevant factor in that these individuals were born between and 1926 and 1930, making them children during the Great Depression, a period of chronic material deprivation for most families (Rockwell and Elder, 1982). It is conceivable that such stressors exposed persons of this birth cohort to greater sympathetic nervous system response and chronic exposure to stress-related hormones, creating heightened vulnerability to personal stressors such as familial death.
Interestingly, we did observe a reduction in AD risk associated with adulthood family member deaths among persons in the oldest subgroup, aged 80 years and older. We offer that this association may reflect mortality selection in that, of all persons born in the earliest birth cohort (1895–1915), those who survived to late old age and participated in the study may represent a physically, genetically and psychologically stronger subgroup, generally (Hawkes et al., 2012). Further, of all persons in this birth cohort, those who were additionally exposed to a modest number of family deaths (fewer than five during all of adulthood), and who survived to participate in the study, may represent a subgroup whose adversity resulted in resilience. This subgroup may therefore have been less vulnerable (than “untested” persons who lacked such an opportunity to develop resilience) to all other types of psychosocial and material stressors throughout life (Post, 1992), with a consequential dampening of the physiological stress response linked to greater AD risk.
Limitations and Strengths
The study supports that exposure to the stress of family member death puts one at greater risk of AD. Without animal models, we can only suggest possible neural mechanisms (heightened levels of glucocorticoid exposure), and cannot prove these specific causal mechanisms. Likewise, cohort effects are evident here, but we can only speculate as to the mechanisms involved.
Secondly, our study examines exposure to a specific life events stressor, but does not examine coping styles or adaptability to stress. Many people are resilient and adapt well psychologically, even when faced with hardship. We are not yet able to tell if coping style or successful adaptation and resiliency contributes to the association between death of family members and risk for AD. Future research should examine whether coping styles or other psychosocial factors served as protective factors against AD, or if exposure to family death remains a risk factor, regardless of psychological resiliency.
Our study was largely homogeneous in ethnic and racial composition, which is both a methodological strength and limitation to the research. Exposure and coping with family death may differ by culture, making our study less generalizable to ethnic minority populations. However, the homogeneity of ethnicity in the sample also removes a possible confounding variable by removing the need to disentangle risk by ethnic group. Thus, while this homogeneity somewhat limits our generalizability, it strengthens internal validity.
CONCLUSIONS
In sum, our study suggests that the stress associated with exposure to family member(s) death is associated with elevated risk for AD. We were able to examine these associations by key risk and protective factors, including age of exposure (childhood vs. adult) and birth cohort, as well as controlling for risk factors for AD including APOE genotype. As there is no known cure for AD presently, research on its risk factors serves an important role in identifying and supporting underlying processes involved in disease onset.
Key points.
Persons who experience familial death during adulthood are at greater risk for AD. This effect was found only among the youngest cohort, which suggests that adverse historical contexts determine vulnerability to the effects of psychosocial stress on AD.
Acknowledgments
This research was supported by the National Institutes of Health grants R01-AG031272 and R01- AG011380. Partial support for all data sets within the Utah Population Database (UPDB) was provided by the University of Utah Huntsman Cancer Institute and the Huntsman Cancer Institute Cancer Center Support grant, P30 CA42014 from the National Cancer Institute. Partial salary support for Daniel Hatch was provided by National Institutes of Health grant T32-AG000029.
We acknowledge the contributions of the following individuals whose activities have helped to ensure the success of the project: Cara Brewer, B.A., John C.S. Breitner, M.D., MPH, Tony Calvert, B.S., Carol Leslie, M.S., Michelle McCart, Ronald G. Munger, Ph.D., MPH, Georgiann Sanborn, M.S., Nancy Sassano, Ph.D., Sarah Schwartz, M.S., Martin Toohill, Ph.D., Heidi Wengreen, Ph.D., RD, James Wyatt, Bonita W. Wyse, Ph.D., and Peter P. Zandi, Ph.D., M.P.H.
Footnotes
CONFLICTS OF INTEREST
None declared by all authors.
References
- Alzheimer’s Association. [Accessed April 1 2014];Alzheimer’s disease facts and figures. 2012 [Online]. Available: http://www.alz.org/downloads/facts_figures_2012.pdf.
- American Psychiatric Association. Diagnostic and Statisical Manual of Mental Disorders: Text Revision. Washington D.C: 1987. [Google Scholar]
- Braak H, Braak E. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiology of Aging. 1997;18:351–357. doi: 10.1016/s0197-4580(97)00056-0. [DOI] [PubMed] [Google Scholar]
- Breitner JCS, Wyse BW, Anthony JC, et al. APOE-ε4 count predicts age when prevalence of AD increases, then declines: The Cache County Study. Neurology. 1999;53:321–331. doi: 10.1212/WNL.53.2.321. [DOI] [PubMed] [Google Scholar]
- Buckley T, Bartrop R, Mckinley S, et al. Prospective study of early bereavement on psychological and behavioural cardiac risk factors. Internal Medicine Journal. 2009;39:370–378. doi: 10.1111/j.1445-5994.2008.01879.x. [DOI] [PubMed] [Google Scholar]
- Elder GH. Children of the Great Depression: Social change in life experience. Chicago: University of Chicago Press; 1974. [Google Scholar]
- Fratiglioni L, Wang HX, Ericsson K, Maytan M, Winblad B. Influence of social network on occurrence of dementia: A community-based longitudinal study. Lancet. 2000;355:1315–1319. doi: 10.1016/s0140-6736(00)02113-9. [DOI] [PubMed] [Google Scholar]
- Greene D, Tschanz JT, Smith KR, et al. Impact of offspring death on cognitive health in late life: The Cache County Study. The American Journal of Geriatric Psychiatry. 2014;22:1307–1315. doi: 10.1016/j.jagp.2013.05.002. doi: http://dx.doi.org/10.1016/j.jagp.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatch DJ, Schwartz S, Norton MC. Depression and antidepressant use moderate association between widowhood and Alzheimer’s disease. International Journal of Geriatric Psychiatry. 2015;30:292–299. doi: 10.1002/gps.4140. [DOI] [PubMed] [Google Scholar]
- Hawkes K, Smith KR, Blevins JK. Human actuarial aging increases faster when background death rates are lower: A consequence of differential heterogeneity? Evolution. 2012;66:103–114. doi: 10.1111/j.1558-5646.2011.01414.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorm AF, Jacomb PA. The Informant Questionnaire on Cognitive Decline in the Elderly (IQCODE) - Socio-demographic correlates, reliability, validity and some norms. Psychological Medicine. 1989;19:1015–1022. doi: 10.1017/S0033291700005742. [DOI] [PubMed] [Google Scholar]
- Leventopoulos M, Ruedi-Bettschen D, Knuesel I, et al. Long-term effects of early life deprivation on brain glia in Fischer rats. Brain Research. 2007;1142:119–126. doi: 10.1016/j.brainres.2007.01.039. [DOI] [PubMed] [Google Scholar]
- Li J, Precht DH, Mortensen PB, Olsen J. Mortality in parents after death of a child in Denmark: A nationwide follow-up study. Lancet. 2003;361:363–367. doi: 10.1016/s0140-6736(03)12387-2. [DOI] [PubMed] [Google Scholar]
- Lloyd C, Smith J, Weinger K. Stress and diabetes: A review of the links. Diabetes Spectrum. 2005;18:121–127. doi: 10.2337/diaspect.18.2.121. [DOI] [Google Scholar]
- Lyons DM, Parker KJ, Katz M, Schatzberg AF. Developmental cascades linking stress inoculation, arousal regulation, and resilience. Frontiers in Behavioral Neuroscience. 2009;3:1–6. doi: 10.3389/neuro.08.032.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mcewen BS. Sex, stress and the hippocampus: Allostasis, allostatic load and the aging process. Neurobiology of Aging. 2002;23:921–939. doi: 10.1016/s0197-4580(02)00027-1. [DOI] [PubMed] [Google Scholar]
- Mckhann G, Drachman D, Folstein M, et al. Clinical-diagnosis of alzheimers-disease - report of the NINCDS-ADRDA work group under the auspices of Department-of-Health-and-Human-Services Task-Force on Alzheimers-disease. Neurology. 1984;34:939–944. doi: 10.1212/WNL.34.7.939. [DOI] [PubMed] [Google Scholar]
- Meaney MJ, Aitken DH, Vanberkel C, Bhatnagar S, Sapolsky RM. Effect of neonatal handling on age-related impairments associated with the hippocampus. Science. 1988;239:766–768. doi: 10.1126/science.3340858. [DOI] [PubMed] [Google Scholar]
- Nam CB, Powers MG. The socioeconomic approach to status measurement. Houston: Cap and Gown Press; 1983. [Google Scholar]
- Norton MC, Breitner JCS, Welsh KA, Wyse BW. Characteristics of nonresponders in a community survey of the elderly. Journal of the American Geriatrics Society. 1994;42:1252–1256. doi: 10.1111/j.1532-5415.1994.tb06506.x. doi: http://dx.doi.org/10.1111/j.1532-5415.1994.tb06506.x. [DOI] [PubMed] [Google Scholar]
- Norton MC, Smith KR, Østbye T, et al. Early parental death and remarriage of widowed parents as risk factors for Alzheimer’s disease. American Journal of Geriatric Psychiatry. 2011;19:814–824. doi: 10.1097/JGP.0b013e3182011b38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oomen CA, Soeters H, Audureau N, et al. Severe early life stress hampers spatial learning and neurogenesis, but improves hippocampal synaptic plasticity and emotional learning under high-stress conditions in adulthood. Journal of Neuroscience. 2010;30:6635–6645. doi: 10.1523/jneurosci.0247-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson G, Skoog I. A prospective population study of psychosocial risk factors for late onset dementia. International Journal of Geriatric Psychiatry. 1996;11:15–22. doi: 10.1002/(sici)1099-1166(199601)11:1<15::aid-gps262>3.3.co;2-x. [DOI] [Google Scholar]
- Post RM. Transduction of psychosocial stress into the neurobiology of recurrent affective-disorder. American Journal of Psychiatry. 1992;149:999–1010. doi: 10.1176/ajp.149.8.999. doi: http://dx.doi.org/10.1176/ajp.149.8.999. [DOI] [PubMed] [Google Scholar]
- Ravona-Springer R, Beeri MS, Goldbourt U. Younger age at crisis following parental death in male children and adolescents is associated with higher risk for dementia at old age. Alzheimer Disease & Associated Disorders. 2012;26:68–73. doi: 10.1097/WAD.0b013e3182191f86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockwell RC, Elder GH. Economic deprivation and problem behavior - childhood and adolescence in the great-depression. Human Development. 1982;25:57–64. doi: http://dx.doi.org/10.1159/000137724. [PubMed] [Google Scholar]
- Roman GC, Tatemichi TK, Erkinjuntti T, et al. Vascular dementia - diagnostic-criteria for research studies - report of the NINDS-AIREN international workshop. Neurology. 1993;43:250–260. doi: 10.1212/WNL.43.2.250. [DOI] [PubMed] [Google Scholar]
- Saunders AM, Strittmatter WJ, Schmechel D, StGeorge-Hyslop PH, Pericak-Vance MA, Joo SH, Roses AD. Association of apolipoprotein E allele ε4 with late-onset familial and sporadic Alzheimer’s disease. Neurology. 1993;43:1467–1472. doi: 10.1212/WNL.43.8.1467. [DOI] [PubMed] [Google Scholar]
- Song JW, Chung KC. Observational studies: Cohort and case-control studies. Plastic and Reconstructive Surgery. 2010;126:2234–2242. doi: 10.1097/PRS.0b013e3181f44abc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennant C. Life events, stress and depression: A review of recent findings. Australian and New Zealand Journal of Psychiatry. 2002;36:173–182. doi: 10.1046/j.1440-1614.2002.01007.x. [DOI] [PubMed] [Google Scholar]
- Tschanz JT, Welsh-Bohmer KA, Plassman BL, et al. An adaptation of the Modified Mini-Mental State examination: Analysis of demographic influences and normative data - The Cache County study. Neuropsychiatry Neuropsychology and Behavioral Neurology. 2002;15:28–38. [PubMed] [Google Scholar]
- Tsolaki M, Papaliagkas V, Kounti F, et al. Severely stressful events and dementia: A study of an elderly Greek demented population. Psychiatry Research. 2010;176:51–54. doi: 10.1016/j.psychres.2009.06.001. [DOI] [PubMed] [Google Scholar]
- Uher R, Mcguffin P. The moderation by the serotonin transporter gene of environmental adversity in the etiology of depression: 2009 update. Molecular Psychiatry. 2010;15:18–22. doi: 10.1038/mp.2009.123. [DOI] [PubMed] [Google Scholar]
- Van Gelder BM, Tijhuis M, Kalmijn S, et al. Marital status and living situation during a 5-year period are associated with a subsequent 10-year cognitive decline in older men: The FINE study. Journals of Gerontology Series B-Psychological Sciences and Social Sciences. 2006;61:P213–P219. doi: 10.1093/geronb/61.4.p213. doi: http://dx.doi.org/10.1093/geronb/61.4.P213. [DOI] [PubMed] [Google Scholar]
- Vitaliano PP, Scanlan JM, Zhang JP, et al. A path model of chronic stress, the metabolic syndrome, and coronary heart disease. Psychosomatic Medicine. 2002;64:418–435. doi: 10.1097/00006842-200205000-00006. doi: http://dx.doi.org/10.1097/00006842-200205000-00006. [DOI] [PubMed] [Google Scholar]
- Whalley LJ, Staff RT, Murray AD, Deary IJ, Starr JM. Genetic and environmental factors in late onset dementia: Possible role for early parental death. International Journal of Geriatric Psychiatry. 2013;28:75–81. doi: 10.1002/gps.3792. [DOI] [PubMed] [Google Scholar]
- World Health Organization. [Accessed April 1 2014];New WHOpenness report: Dementia: A public health priority. 2012 [Online]. Available: http://www.who.int/mediacentre/news/releases/2012/dementia_20120411/en/index.html.
- Yang Y. Is old age depressing? Growth trajectories and cohort variations in late-life depression. Journal of Health and Social Behavior. 2007;48:16–32. doi: 10.1177/002214650704800102. doi: http://dx.doi.org/10.1177/002214650704800102. [DOI] [PubMed] [Google Scholar]