

Abstract
Enzymes use protein architecture to impose specific electrostatic fields onto their bound substrates, but the magnitude and catalytic effect of these electric fields have proven difficult to quantify with standard experimental approaches. Using vibrational Stark effect spectroscopy, we found that the active site of the enzyme ketosteroid isomerase (KSI) exerts an extremely large electric field onto the C=O chemical bond that undergoes a charge rearrangement in KSI’s rate-determining step. Moreover, we found that the magnitude of the electric field exerted by the active site strongly correlates with the enzyme’s catalytic rate enhancement, enabling us to quantify the fraction of the catalytic effect that is electrostatic in origin. The measurements described here may help explain the role of electrostatics in many other enzymes and biomolecular systems.
Ketosteroid isomerase (KSI) is a small, proficient enzyme with one of the highest known unimolecular rate constants in biochemistry (1, 2), which has prompted extensive study of its mechanism and the catalytic strategies it uses (3–5). In steroid biosynthesis and degradation, KSI alters the position of a C=C double bond (Fig. 1A) by first abstracting a nearby a proton (E•S ⇌ E•I), forming a charged enolate intermediate (E•I), and then reinserting the proton onto the steroid two carbons away (E•I ⇌ E•P). The removal of a proton in the first step initiates a rehybridization that converts the adjacent ketone group to a charged enolate, an unstable species that is normally high in free energy and so slow to form. The reaction is therefore expected to produce an increase in dipole moment at the carbonyl bond ( ), suggesting that KSI may facilitate this reaction by exerting an electric field ( ) on this bond that stabilizes it in the intermediate form and the preceding transition state (Fig. 1B). Using vibrational Stark effects, we have measured the electric field that KSI exerts on this C=O bond, providing quantitative experimental evidence for the connection between electrostatics and catalytic proficiency.
Fig. 1. Catalysis by ketosteroid isomerase.
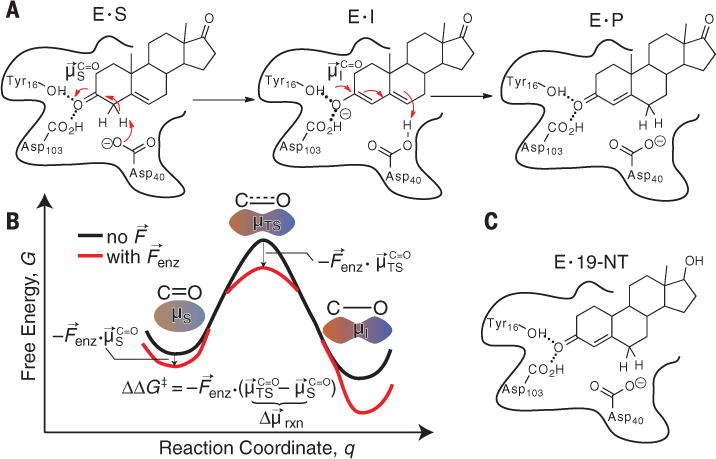
(A) The chemical mechanism of ketosteroid isomerase. In the first step, Asp40 removes an a proton from the steroid to form an enolate, stabilized by two H-bonds from Tyr16 and Asp103. This transformation results in an increase of the dipole moment along the C=O bond. E, enzyme; S, substrate; I, intermediate; P, product. (B) Simple model for electrostatic catalysis, illustrating the effect of an electric field from the organized environment of an enzyme active site ( ) on a reaction’s activation barrier (ΔG‡). , dipole of substrate’s C=O bond; , dipole of transition state’s C=O bond. (C) Complex between KSI and the product-like inhibitor, 19-NT, used in this study to probe electric fields in the KSI active site.
The frequencies of certain vibrations (such as the C=O stretch) shift in a linear manner with the electric field experienced by that vibration from its environment, a phenomenon known as the linear vibrational Stark effect (6, 7). Through this effect, we have shown that vibrations can be used as probes of local electrostatic fields. The nitrile group has been widely deployed to measure electric fields inside enzymes and their relationship to mutation (8), ligand occupancy (9), or conformational changes over the catalytic cycle (10). In this study, we have focused on the C=O group of the inhibitor 19-nortestosterone (19-NT) (Fig. 1C), because when 19-NT binds, the C=O group is loaded directly into the catalytic machinery (11, 12). In this way, 19-NT’s C=O vibrational (infrared) frequency shift probes the electrostatic environment that the substrate’s C=O bond would experience in the active site, except 19-NT cannot react due to the position of the C=C bond.
To calibrate the sensitivity of 19-NT’s C=O vibrational frequency to an electric field, we used two complementary approaches. In Stark spectroscopy (Fig. 2, A and B), an external electric field of known magnitude is applied to a frozen glass containing 19-NT, and the accompanying effect on the vibrational spectrum is recorded (7). By fitting the Stark spectrum (Fig. 2B) to derivatives of the absorption spectrum (Fig. 2A), the vibration’s difference dipole can be extracted: , where f is the local field factor (fig. S1) (6, 7, 13). A vibration’s difference dipole is its linear Stark tuning rate; that is, 19-NT’s C=O vibrational frequency shifts ~1.4/f cm−1 for every MV/cm of electric field projected onto the C=O bond axis, whether the source of that field is an external voltage (as in Stark spectroscopy) or an organized environment created by an enzyme active site ( ) that we wish to characterize. Whenever an external field is applied to a vitreous sample, vibrational bands will broaden because 19-NT molecules (and their C=O bonds) are randomly oriented with respect to the fixed direction of the external electric field (6, 7). By contrast, a vibrational probe will have a fixed orientation with respect to a protein electric field when bound to a protein, and as such the linear Stark effect then produces spectral shifts instead of broadening. The C=O vibration’s Stark tuning rate does not appreciably change when C=O accepts a hydrogen bond (fig. S2), implying that the frequency still responds to fields linearly even when C=O participates in stronger interactions, although those interactions themselves are associated with larger electric fields (14).
Fig. 2. Sensitivity of the C=O stretch frequency of 19-NT to electrostatic field.

(A) The absorption spectrum of 19-NT (50 mM) in glassy 2-methyl tetrahydrofuran at 77 K. (B) The Stark spectrum of 19-NT at 1.0 MV/cm, shown as a difference between the field-on and field-off spectra. The Stark tuning rate is related to the second derivative fitting parameter (figs. S1 and S2). (C) Infrared spectra of 19-NT dissolved in organic solvents of various polarities or water; the small peak at 1615 cm−1 is the C=C stretch. (D) Plot of 19-NT’s C=O peak frequency, against the calculated solvent electric field, , the C=O group experiences in each of those solvents (13, 14). The least-squares regression line is (R2 = 0.99). Error ranges for frequencies are contained within symbols; for electric fields, error bars report the correlation-adjusted error of the average electric field from 2-ns simulations.
We also pursued a second approach to calibrate the sensitivity of 19-NT’s C=O frequency shifts to electric fields and to assign these frequencies to an absolute field scale: Specifically, we measured 19-NT’s vibrational spectrum in a series of solvents (table S1) and examined the correlation of the frequencies with the solvents’ reaction fields, estimated by molecular dynamics (MD) simulations (14). As shown in Fig. 2C, 19-NT’s C=O band shifts consistently to the red with increasing solvent polarity, from 1690.2 cm−1 in nonpolar hexane to 1634.0 cm−1 in water, so that the C=O frequency shifts across a 56 cm−1 span from solvatochromic effects. The large redshift in water reflects the large electric fields that are created by water’s hydrogen bonds (H-bonds). The trend in solvent shifts is strongly correlated with the average electric field that each solvent exerts on the C=O bond (Fig. 2D and fig. S3A); that is, the plot of observed frequency versus computed field is linear (R2 = 0.99) and its slope corroborates the Stark tuning rate measured by Stark spectroscopy. The ~2-fold difference between the slope of this curve [0.702 cm−1/(MV/cm)] and the measured Stark tuning rate is consistent with the current understanding of the local field effect (f ~ 2), based on other vibrational probes and electrostatic models (text S1) (13, 14). The regression line implies that the frequency variation due to different molecular environments can be well explained as a field effect and suggests that we can model 19-NT’s C=O peak frequency in terms of the average electric field experienced by the vibration.
When 19-NT is bound to wild-type KSI, the C=O probe engages in short, strong H-bonds with Tyr16 and Asp103 (11, 12), and its vibrational frequency reflects the electric field at a primary site of charge rearrangement during KSI’s catalytic cycle. Notably, the C=O vibration red-shifts to 1588.3 cm−1 (Fig. 3A), 46 cm−1 further to the red from the peak frequency in water, implying an extremely large electrostatic field. Attributing the frequency shift to the Stark effect, the linear field-frequency relationship of Fig. 2D maps this frequency value to an ensemble-average electric field of −144 ± 6 MV/cm. Although this highly red-shifted frequency lies outside the known linear range from solvatochromism, additional lines of evidence suggest that the C=O vibrational frequency maintains an approximately linear relationship with the field in this regime; neglect of higher-order terms is expected to result in overestimates of the electric field, but by no more than 10% (fig. S4 and text S2). Not only is the C=O band extremely red-shifted in KSI, it is also extremely narrow (Fig. 3A), suggesting a rather rigid environment (15) that greatly reduces the dispersion in the electric field. This is very different from what is observed in H-bonding solvents like water that exert large, but also highly inhomogeneous, electric fields because solvent H-bonds can assume a broad distribution of conformations (dashed traces in Fig. 3A and fig. S3, B and C) (14). Furthermore, the position of the C=O band in wild-type KSI is situated at the reddest (highest field) edge of the frequencies sampled by the C=O group in water (see the red and dashed traces in Fig. 3A), suggesting that the active site achieves this large field by restricting H-bond conformations to those that are associated with the largest electric fields.
Fig. 3. Contribution of active-site electric fields to KSI’s catalytic effect.

(A) Infrared spectra of 19-NT bound to the active site of wild-type and mutant KSI (color traces), shown alongside a few spectra of 19-NT in solvents (dotted black traces) as a reference. DMSO, methyl sulfoxide; WT, wild type. (B) Plot of enzymatic unimolecular free energy barrier, ΔG‡, against the electric field, , 19-NT’s C=O experiences in of group experiences in each the KSI active sites. Expressing ΔG‡ in kcal mol−1 and in units of kcal mol−1D−1 (upper axis), the least-squares regression line is . Error ranges for free energy barriers are contained within symbols; for electric fields, error bars report both experimental error from vibrational frequency measurements and model error from mapping frequencies to electric fields. (C) Relative contribution of KSI’s catalytic strategies (electric field effect and general base positioning) to speed up KSI’s reaction relative to its rate in solution.
By exploring a series of structurally conservative (but catalytically detrimental) mutants (table S2), we could systematically perturb the catalytic efficacy of KSI and quantitatively evaluate its relationship to the electric field probed by the C=O vibration. In all cases, the assignment of the vibrational bands to 19-NT was confirmed with isotope replacement studies using C=18O 19-NT (figs. S5 and S6). The H-bond provided by Tyr16 is known to be essential for KSI’s catalysis, as the conservative Tyr16Phe mutation diminishes KSI’s rate by factors of ~104 (11, 16). This single point mutation induced a blue shift from 1588.3 cm−1 to 1647.5 cm−1 (Fig. 3A), implying a much smaller average electric field. (This change in field magnitude is comparable to that of the change in solvent field between hexane and water.) The Tyr16Ser mutation (17), although less conservative than the Phe substitution, is actually less detrimental. This observation has been explained by the suggestion that leaving a cavity in Tyr16’s place allows water to remain in the back pocket of the active site; these water molecules could also H-bond to the steroid substrate, thereby partially compensating for the loss of Tyr16’s key H-bond (18). Indeed, the C=O stretching frequency in Tyr16Ser is not as dramatically blue-shifted as it is in Tyr16Phe. Asp103 is the other primary H-bond donor in KSI’s active site (Fig. 1A). In the Asp103Asn mutant, the H-bonding proton is much less acidic, but N–H and O–H bonds have similar sterics and dipole moments. Consistent with these considerations, the change in electric field and the rate impairment this mutation entails with respect to wild-type are much smaller compared to the other mutants.
Although it is conventionally accepted that KSI uses Tyr16’s and Asp103’s H-bonds to stabilize its transition state (1, 11), these measurements show that the interaction between these H-bonds and the C=O group can be described fundamentally in terms of the electric field they produce. In all the mutants studied, 19-NT’s C=O band remains relatively narrow, suggesting that electrostatic rigidity is conferred by KSI’s scaffold, rather than by the H-bonding residues. Also, sizable electric fields persist in the KSI active site after removing the critical H-bond donated by Tyr16 (−60 ± 3 MV/cm), implying that a substantial electrostatic field contribution also arises from the environment fashioned by the enzyme scaffold (Fig. 3B). Nevertheless, that the Tyr16 H-bond alone contributes a static field of 84 ± 7 MV/cm without an accompanying increase in electric field dispersion makes it quite distinct from water, which donates close to two H-bonds to C=O on average, but taken altogether these H-bonds generate an average field of ~40 MV/cm that is also highly heterogeneous (13).
The Asp40Asn mutant decreases KSI’s catalytic rate by a factor of ~106 (16) but only blue-shifts the C=O vibrational frequency of bound 19-NT by 6 cm−1 relative to wild-type KSI (table S2). This behavior strongly contrasts with other mutants studied, which produce blue-shifts commensurate with their deleterious effect on catalysis (Fig. 3B). To explain this observation, we posit that, whereas the function of Tyr16 and Asp103 is to stabilize the reaction’s transition state by exerting electrostatic fields onto the carbonyl moiety, Asp40 provides an orthogonal catalytic function (text S3), by acting as the general base in the proton-transfer reaction (Fig. 1A).
When nitrile vibrational probes were placed in other locations around the KSI active site in previous work, frequencies were found well within the range demarcated by the solvent series (9). In other words, the extreme electric field experienced by 19-NT’s C=O in wild-type KSI is specific to its precise position in the active site where strong local interactions and the collective effect of the overall enzyme architecture mutually reinforce each other.
A plot of each mutant’s apparent activation barrier (calculated from the Michaelis-Menten kcat) (fig. S7A) (16, 18, 19) against its corresponding ensemble-average active-site electric field magnitude (derived from the field-frequency calibration curve in Fig. 2D) reveals a robust linear trend (Fig. 3B; see also fig. S7B). This relationship suggests that electric fields in KSI’s active site are intimately linked to catalysis. Moreover, this correlation can be explained using the simple model for electrostatic catalysis in Fig. 1B. The formation of a transition state generally involves redistribution of electron density, resulting in bonds with larger dipole moments than the analogous bonds in the substrate (e.g., ) (20, 21). An electric field will therefore differentially stabilize the transition state in proportion to the reaction difference dipole ( ), altering the activation barrier by (Fig. 1B). If we make a simplifying assumption that the electric field experienced by the C=O bond is the same in the substrate and transition state, the model can be directly mapped onto the data in Fig. 3B; the slope of the plot (1 D) corresponds to , and the intercept (18.8 kcal mol−1) corresponds to the hypothetical activation barrier if KSI contributed no stabilizing electric field. The small value for implies that there a rather small perturbation in j the electrostatic character of the substrate upon activation, consistent with ab initio density functional theory calculations (fig. S7C and table S3). Apparently, the very large field present in the KSI active site is needed to leverage what small charge displacement is associated with the reaction’s transition state. A chemical reaction with a larger charge displacement would consequently be exponentially more accelerated by these electric fields, suggesting that electric field effects may provide a natural framework for explaining the catalysis of more proficient enzymes as well (22).
By comparing the intercept extrapolated to from Fig. 3B (18.8 kcal mol−1) to wild-type KSI’s activation barrier (11.5 kcal mol−1), we estimate that the active site’s electric field contributes 7.3 T 0.4 kcal mol−1 to KSI’s barrier reduction. This corresponds to an ~105-fold rate enhancement and accounts for 70% of KSI’s catalytic speedup relative to an uncatalyzed reference reaction in solution (Fig. 3C and text S3). Moreover, the electrostatic stabilization energy is quite similar to the enthalpic component of KSI’s barrier reduction (9 kcal mol−1), suggesting that the thermodynamic manifestation of the electric field effect is enthalpic (23). The active site’s suppression of electric field variability, as evidenced by the approximately lifetime-limited linewidth of the C=O vibrational band in all the KSI mutants, is possibly also an important catalytic feature; for example, the Tyr16Phe mutant produces a smaller average electric field than water (Fig. 3A), although it is still a modest catalyst relative to solution. However, the linear infrared experiments described here cannot directly quantitate this effect.
The remaining catalytic effect beyond electrostatics can likely be attributed to the precise positioning of the general base (Asp40) with respect to the proton on the steroid to be abstracted, which is expected to be an entropic effect. From this discussion, we surmise that in enzymatic proton abstraction, electrostatic stabilization and exact positioning of reacting moieties provide the physical basis to achieve enthalpic and entropic contributions to catalysis, respectively; however, contrary to earlier views (24, 25), electrostatic stabilization can be the more important of the two (Fig. 3C).
What is the physical basis for the extreme electric field detected in KSI’s active site? Large electric fields arising from Tyr16 and Asp103 are expected when the carbonyl group of the ligand closely approaches the OH groups of these two residues and in a coplanar orientation [as indeed is seen in crystal (3) and solution (12) structures]. Nevertheless, these static structures cannot predict or reproduce the electric fields determined by vibrational Stark effects, likely because structures represent ensemble averages and because electric fields depend sensitively on atomic positions down to resolutions not accessible in most structural data (text S4). Moreover, electric fields calculated from classical MD simulations of the KSI•19-NT complex (text S4) also do not agree with our experiments (text S4 and fig. S8). Better estimates of active-site electric fields have been obtained with more sophisticated computational models, which have provided a theoretical foundation supporting the link between electric fields and catalysis (10, 21).
Unusual spectral shifts have been observed previously in enzyme active sites (including KSI) (11, 26–28) and have been variously interpreted as implying strain, distortion, or polarization. In contrast, the vibrational Stark effect enables a quantitative connection between spectroscopic observables and a fundamental physical quantity (electric field). As we demonstrate here, these experiments can be applied to measuring the H-bond electric field strength in proteins in functional contexts. Moreover, the electric field description provides a framework to quantify the contributions of specific H-bonding interactions, as well as the overall electrostatic environment with the same units, to give a simple and unified model for electrostatic catalysis (Fig. 1B). That a substantial portion of KSI’s catalytic rate enhancement can be explained in terms of its average electric field suggests that the electric field could be a useful design criterion in the ongoing efforts to engineer enzymes with unnatural or enhanced functions. More generally, we anticipate that electric field measurements with functionally relevant vibrational probes will elucidate the physical basis for a broad spectrum of biomolecular and condensed phase interactions and processes.
Supplementary Material
Acknowledgments
We thank the laboratory of D. Herschlag for providing plasmids of some of the KSI mutants. S.D.F thanks the NSF predoctoral fellowship program and the Stanford Bio-X interdisciplinary graduate fellowship for support. This work was supported in part by a grant from NIH (grant GM27738). S.D.F. designed the study. S.D.F. and S.B. performed experiments and collected data. S.D.F. analyzed the data. S.D.F. and S.G.B. discussed results and wrote the manuscript.
Footnotes
SUPPLEMENTARY MATERIALS
www.sciencemag.org/content/346/6216/1510/suppl/DC1 Materials and Methods
References (29–70)
REFERENCES AND NOTES
- 1.Pollack RM. Bioorg Chem. 2004;32:341–353. doi: 10.1016/j.bioorg.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 2.Hawkinson DC, Eames TC, Pollack RM. Biochemistry. 1991;30:10849–10858. doi: 10.1021/bi00109a007. [DOI] [PubMed] [Google Scholar]
- 3.Kraut DA, et al. PLOS Biol. 2006;4:e99. doi: 10.1371/journal.pbio.0040099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kamerlin SCL, Sharma PK, Chu ZT, Warshel A. Proc Natl Acad Sci USA. 2010;107:4075–4080. doi: 10.1073/pnas.0914579107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Herschlag D, Natarajan A. Biochemistry. 2013;52:2050–2067. doi: 10.1021/bi4000113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boxer SG. J Phys Chem B. 2009;113:2972–2983. doi: 10.1021/jp8067393. [DOI] [PubMed] [Google Scholar]
- 7.Andrews SS, Boxer SG. J Phys Chem A. 2000;104:11853–11863. [Google Scholar]
- 8.Stafford AJ, Walker DM, Webb LJ. Biochemistry. 2012;51:2757–2767. doi: 10.1021/bi201225p. [DOI] [PubMed] [Google Scholar]
- 9.Fafarman AT, Sigala PA, Herschlag D, Boxer SG. J Am Chem Soc. 2010;132:12811–12813. doi: 10.1021/ja104573b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu CT, et al. J Am Chem Soc. 2014;136:10349–10360. doi: 10.1021/ja5038947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuliopulos A, Mildvan AS, Shortle D, Talalay P. Biochemistry. 1989;28:149–159. doi: 10.1021/bi00427a022. [DOI] [PubMed] [Google Scholar]
- 12.Massiah MA, Abeygunawardana C, Gittis AG, Mildvan AS. Biochemistry. 1998;37:14701–14712. doi: 10.1021/bi981447b. [DOI] [PubMed] [Google Scholar]
- 13.Fried SD, Wang LP, Boxer SG, Ren P, Pande VS. J Phys Chem B. 2013;117:16236–16248. doi: 10.1021/jp410720y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fried SD, Bagchi S, Boxer SG. J Am Chem Soc. 2013;135:11181–11192. doi: 10.1021/ja403917z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Childs W, Boxer SG. J Am Chem Soc. 2010;132:6474–6480. doi: 10.1021/ja1007849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim SW, Choi KY. J Bacteriol. 1995;177:2602–2605. doi: 10.1128/jb.177.9.2602-2605.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nam GH, et al. Biochemistry. 2001;40:13529–13537. doi: 10.1021/bi015547k. [DOI] [PubMed] [Google Scholar]
- 18.Kraut DA, Sigala PA, Fenn TD, Herschlag D. Proc Natl Acad Sci USA. 2010;107:1960–1965. doi: 10.1073/pnas.0911168107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Choi G, et al. Biochemistry. 2001;40:6828–6835. doi: 10.1021/bi002767+. [DOI] [PubMed] [Google Scholar]
- 20.Warshel A. Proc Natl Acad Sci USA. 1978;75:5250–5254. doi: 10.1073/pnas.75.11.5250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Warshel A, et al. Chem Rev. 2006;106:3210–3235. doi: 10.1021/cr0503106. [DOI] [PubMed] [Google Scholar]
- 22.Radzicka A, Wolfenden R. Science. 1995;267:90–93. doi: 10.1126/science.7809611. [DOI] [PubMed] [Google Scholar]
- 23.Houck WJ, Pollack RM. J Am Chem Soc. 2004;126:16416–16425. doi: 10.1021/ja046819k. [DOI] [PubMed] [Google Scholar]
- 24.Page MI, Jencks WP. Proc Natl Acad Sci USA. 1971;68:1678–1683. doi: 10.1073/pnas.68.8.1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Knowles JR. Nature. 1991;350:121–124. doi: 10.1038/350121a0. [DOI] [PubMed] [Google Scholar]
- 26.Austin JC, Spiro TG, Kuliopulos A, Mildvan AS. Protein Sci. 1992;1:259–270. doi: 10.1002/pro.5560010208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carey PR, Tonge PJ. Acc Chem Res. 1995;28:8–13. [Google Scholar]
- 28.Belasco JG, Knowles JR. Biochemistry. 1980;19:472–477. doi: 10.1021/bi00544a012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.