

Abstract
UvrD helicase is required for nucleotide excision repair, although its role in this process is not well defined. Here we show that Escherichia coli UvrD binds RNA polymerase during transcription elongation and, using its helicase/translocase activity, forces RNA polymerase to slide backward along DNA. By inducing backtracking, UvrD exposes DNA lesions shielded by blocked RNA polymerase, allowing nucleotide excision repair enzymes to gain access to sites of damage. Our results establish UvrD as a bona fide transcription elongation factor that contributes to genomic integrity by resolving conflicts between transcription and DNA repair complexes. We further show that the elongation factor NusA cooperates with UvrD in coupling transcription to DNA repair by promoting backtracking and recruiting nucleotide excision repair enzymes to exposed lesions. Because backtracking is a shared feature of all cellular RNA polymerases, we propose that this mechanism enables RNA polymerases to function as global DNA damage scanners in bacteria and eukaryotes.
Nucleotide excision repair (NER) is the most versatile and evolutionarily conserved mechanism used by prokaryotic and eukaryotic cells to repair diverse types of DNA lesions1,2. In bacteria, the general NER pathway commences when UvrA/UvrB proteins bind damaged DNA and recruit UvrC to cleave the impaired strand on both sides of the lesion. The resulting oligonucleotide is displaced by UvrD and/or DNA polymerase I, which fills the gap using the complementary strand as a template2–4.
NER rates are usually greatest at transcriptionally active genes. Moreover, the transcribed DNA strand is preferentially repaired compared to the non-transcribed strand5. This phenomenon, known as transcription-coupled repair (TCR), is a sub-pathway of global NER3,6. The current model of bacterial TCR postulates that a DNA lesion blocking the progression of the transcription elongation complex is shielded from NER enzymes by the stalled RNAP. The DNA translocase, Mfd, binds to the stalled EC through the β subunit of RNA polymerase (RNAP) and dislodges the complex by ‘pushing’ it forward7–10. Concurrently, Mfd recruits UvrA to the exposed lesion site to expedite NER10.
Here we propose an alternative TCR model whose key component is UvrD, a member of DNA helicase superfamily 1, which translocates in a 3′ to 5′ direction using a single-strand, DNA-dependent, ATPase activity11–13. In contrast to Mfd, UvrD facilitates NER by pulling RNAP backward from the DNA lesion without causing termination. Our model further explains the role of elongation factor NusA, which is known to contribute to Mfd-independent TCR14. In this model RNAP recruits the NER complex via UvrD/NusA to the damage site.
UvrD binds RNAP in vitro and in vivo
We performed a mass spectrometry (MS)-assisted survey of proteins that interact with E. coli RNAP in vivo by treating E. coli K12 MG1655 cultures with formaldehyde and isolated RNAP-containing material. Peptides in this material were identified by tandem liquid chromatography mass spectrometry (LC-MS/MS) and used to calculate an exponentially modified protein abundance index (emPAI). This label-free method estimates the relative amount of proteins by the number of sequenced peptides per protein compared with the number of theoretically observable peptides15. UvrD appeared in RNAP crosslinked complexes in abundance comparable to that of bona fide transcription elongation/termination factors NusA, NusG or Rho (Extended Data Fig. 1), indicating a potential direct interaction between UvrD and RNAP.
To verify that UvrD, interacts with RNAP directly, we performed in vitro pull-down assays with purified UvrD and His6-tagged RNAP adsorbed to metal-chelating beads. UvrD bound to the beads only in the presence of immobilized RNAP and remained bound through multiple washings (Extended Data Fig. 1b). The UvrD–RNAP core complex was also isolated in a major discrete peak by size-exclusion chromatography (Extended Data Fig. 1c). Collectively, these data demonstrate stable and specific binding of UvrD to RNAP.
UvrD promotes RNAP backtracking
To investigate the role of UvrD in transcription we reconstituted a single-round runoff assay that measured ‘walking’ (NTP supply-controlled elongation) of the elongation complex along DNA16. We observed little effect of UvrD on RNAP promoter binding and open complex formation (not shown); however, UvrD dramatically influenced elongation, interrupting transcription at many positions along the template, so that only a fraction of elongation complexes produced a full-length (runoff) transcript (Fig. 1a, lane 2). Some UvrD-induced transcriptional ‘arrests’ coincided with pre-existing pause sites, whereas others formed de novo. Most did not change significantly with time, indicating that the corresponding elongation complexes were either permanently arrested or terminated by UvrD. We excluded the latter possibility by showing that the majority of those transcripts remained bound to RNAP after extensive washing with high-salt buffer (Fig. 1a, lane 3).
Figure 1. UvrD promotes RNAP backtracking.

a, EC20 was combined with wild-type UvrD (lanes 2–5), catalytically inactive UvrDE221Q, (lanes 6, 7), or no UvrD (lane 1) before NTP chase (lanes 1, 2, 6) and wash (lanes 3, 7). Red lines connect corresponding RNAs from arrested elongation complexes before and after GreB cleavage (lanes 4, 5). b, EC11 was walked to positions 36, 38 and 39. UvrD was added ± ATP and chased. Inactivated EC36–39 (%) is indicated. c, The p1EC constructs21 (left) and primer extension analyses (right). CAA modifications on the non-template strand of p1EC + pVector (lanes 2, 3) or p1EC + pUvrD (lanes 4, 5). The lac operator (Lac) and transcription bubble are indicated. Red lines show elongation complex position.
Most transcriptional pauses and arrests are caused by backtracking—a reverse sliding of RNAP along DNA and RNA17. In bacteria, backtracked RNAP is prone to transcript cleavage stimulated by Gre factors, which reactivate elongation complexes by removing the extruding 3′ portion of the nascent RNA18,19. To test whether elongation complexes arrested by UvrD were backtracked, we first washed them to remove unincorporated NTPs and UvrD, and incubated with GreB (Fig. 1a, lane 4). GreB shortened most of the transcripts from UvrD-arrested elongation complexes and reactivated these complexes in the presence of NTPs (lane 5), indicating that UvrD induces backtracking at multiple positions during elongation (Extended Data Fig. 2).
To determine whether the enzymatic activity of UvrD was required for the arrest, we repeated the above experiment with UvrDE221Q; the E221Q mutation in the UvrD active site results in ∼600-fold decrease in helicase/translocase activity without significantly affecting binding of DNA, ATP20 or RNAP (not shown). UvrDE221Q failed to cause RNAP arrest during elongation (Fig. 1a, lanes 6, 7).
To confirm the requirement of ATP for UvrD-mediated transcriptional arrest, we walked RNAP to stalled elongation positions EC36–39, washed the beads to remove unincorporated NTPs and incubated the stalled complexes with UvrD ± ATP (Fig. 1b). After 1 min of incubation with UvrD + ATP most of EC36, EC38 and EC39 were inactivated: they failed to resume elongation upon addition of NTPs (lane 15); inactivation was almost complete after 2 min (lane 16). Without UvrD (lanes 2–6) or with UvrD lacking ATP (lanes 8–12), only ∼30% of EC39 was inactivated after 8 min of incubation, whereas EC36 and EC38 remained fully active. We conclude that UvrD induces RNAP backtracking at many positions through its ATP-dependent motor function (Extended Data Fig. 2).
To monitor the effect of UvrD on RNAP backtracking in vivo, we used a plasmid (p1EC) in which RNA synthesis initiated at a constitutive promoter is halted at a downstream position by the lacO-bound Lac repressor (Fig. 1c). The plasmid was designed so that the repressor blocked one isolated elongation complex21. Cells carrying pEC1 were transformed with a UvrD overexpression plasmid (pUvrD) or an empty vector (pVector). To monitor the effect of UvrD and the position of the halted elongation complex we performed in situ footprinting of its DNA bubble using the single-strand-specific probe, chloroacetaldehyde (CAA) (Fig. 1c). The halted elongation complex was clearly backtracked over a longer distance in the presence of pUvrD than empty vector: new CAA reactive sites were detected upstream and the reactivity of the downstream margin of the footprint was decreased (lane 4). Thus, as observed in vitro, UvrD also causes RNAP to backtrack in vivo.
UvrD facilitates NER by towing RNAP
RNAP stalled at DNA lesions presents a major obstacle for NER by obscuring damaged sites from repair enzymes1. The role of UvrD, therefore, could be to clear such lesions by forcing the obstructing elongation complex to backtrack. To test this hypothesis we reconstituted the first steps of NER in vitro (Fig. 2a and Extended Data Fig. 3). An initial elongation complex was assembled on DNA carrying a single cyclobutane pyrimidine dimer CPD) in the template strand at position +57 with respect to the transcriptional start site (Fig. 2a and Extended Data Fig. 3). CPD is the most common ultraviolet (UV)-induced lesion, a substrate for UvrABC and a roadblock for RNAP22–24 (Extended Data Fig. 3). RNAP positioned 46 nucleotides upstream of CPD (EC11) did not affect UvrABC-directed CPD excision (Fig. 2a, lanes 3, 4, 7, 8). However, chasing EC11 to CPD inhibited the UvrC DNA cleavage reaction (lanes 11, 12). Addition of UvrD and ATP restored UvrC endonuclease activity at the CPD site (Fig. 2a, lanes 13–16), indicating that towing RNAP from the lesion site by UvrD is the first step of NER (Fig. 2a).
Figure 2. RNAP backtracking facilitates NER.

a, UvrD pulls RNAP from thymine dimers (TT). The T7A1 promoter template with TT (red) and schematic overview (top); EC11 was chased to TT (lanes 9–16) (bottom). Where indicated, UvrD was added for 5 min. Red boxes show UvrC-mediated DNA cleavage products (19 nucleotides); per cent cleavage is averaged ( ± s.e.m.) from four independent experiments P < 0.05. b, Left, the primer extension experimental overview. Right, primer extension products reflect the location of lesions. Orange lines indicate repair zones for percentage repair. Asterisks indicate representative lesions repaired faster in greAB than in wild-type cells (blue), at the same rate (green) or ‘non-disappearing’ bands for normalization (orange).
Anti-backtracking factors interfere with NER
In the RNAP ‘towing’ model of NER factors that normally inhibit backtracking may hinder the repair process: the GreA and GreB transcript cleavage factors18 and active ribosomes control transcription elongation through this inhibition21. To test this prediction we used a primer extension assay to monitor UV-induced lesion repair in vivo. Such lesions block DNA polymerase, generating truncated primer extension products that can be analysed at single-nucleotide resolution25. We used a plasmid-borne lacZ gene isolated from wild-type and backtracking-prone greA greB mutant cells (Fig. 2b). The primer extension of lacZ DNA taken from both wild-type and mutant cells before and shortly after UV irradiation revealed truncated species unique to the irradiated DNA (Fig. 2b). However, the lesion repair rate (truncated species disappearance) was considerably higher in gre− cells than in wild-type cells. Only a few lesions were repaired at the same rate in wild-type and Gre-deficient cells. Because this assay detects any DNA damage that halts Taq DNA polymerase (not only pyrimidine dimers) we conclude that: (1) most UV-induced adducts within the transcribed region block the progression of RNAP, hampering the repair process, and (2) RNAP backtracking facilitates repair at most of damage sites.
If these conclusions are correct, GreA/GreB deficiency should suppress UvrD sensitivity to UV and genotoxic agents against which NER provides protection. We examined three genotoxic chemicals: mitomycin C, 4-nitroquinoline-1-oxide (4NQO) and cisplatin. These chemotherapeutics generate crosslinks and bulky DNA adducts that are predominantly processed by NER26. uvrD cells were highly sensitive to killing by all three agents, whereas inactivation of greAB greatly suppressed uvrD sensitivity to all three agents (Fig. 3a and Extended Data Fig. 4) and to UV irradiation (Fig. 3b and Extended Data Fig. 4c). As the function of GreAB is to suppress RNAP backtracking, we conclude that RNAP backtracking is required for efficient NER in vivo and that UvrD is responsible for much of the NER-associated RNAP backtracking during genotoxic stress.
Figure 3. Anti-backtracking factors obstruct UvrD activity in NER.
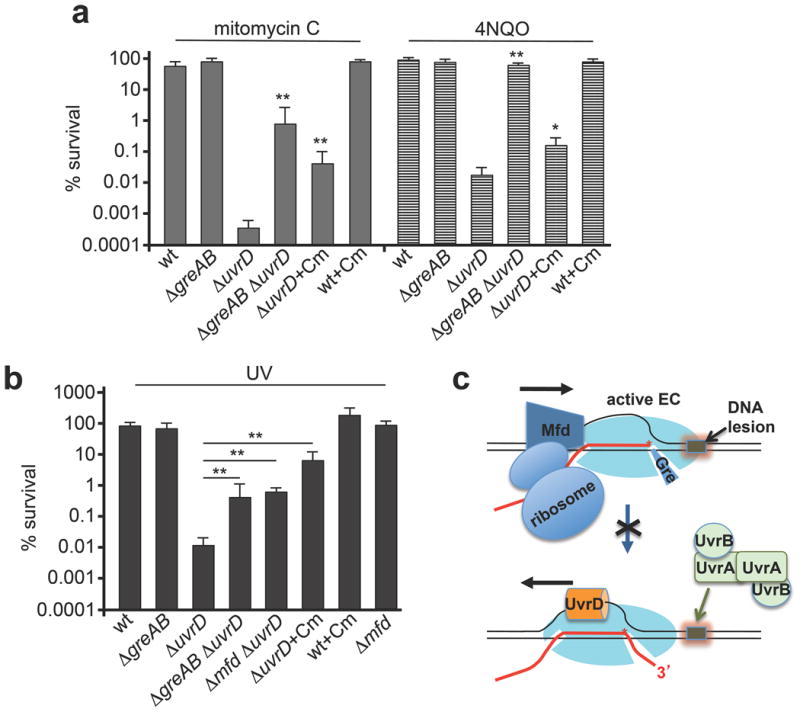
a, Inactivating greAB or slowing ribosomal translocation (with 1 μg ml−1 chloramphenicol) suppresses uvrD sensitivity to mitomycin C (1 μg ml−1; filled bars) and 4NQO (1 μM; striped bars). Data from three independent experiments are presented as the mean ± s.e.m.; *P < 0.05, **P < 0.01. Cm, chloramphenicol; wt, wild type. b, Inactivating greAB, mfd or slowing ribosomal translocation suppresses uvrD sensitivity to UV irradiation (5 J per m2 at 30 °C). Data from three independent experiments are presented as the mean ± s.e.m.; **P < 0.01. c, Cartoon summarizing interference with NER.
In bacteria, active ribosomes control the rate of transcription elongation at protein coding sequences by ‘pushing’ RNAP forward21, whereas inhibited ribosomes render cells highly resistant to UV-mediated lethality27,28. To test if active ribosomes interfere with NER by decreasing the frequency of backtracking we used a sublethal dose of chloramphenicol to slow ribosomes' translocation21. Analogous to the situation with Gre-deficiency, chloramphenicol rendered uvrD cells more resistant to 4NQO, mitomycin (Fig. 3a and Extended Data Fig. 4b) and UV (Fig. 3b and Extended Data Fig. 4d).
Collectively, these results argue that UvrD competes with anti-backtracking factors during genotoxic stress to promote NER (Fig. 3c). Interestingly, UvrD counteracts GreAB even under normal growth conditions, because uvrD inactivation suppresses the temperature sensitivity of gre− cells (Extended Data Fig. 5).
Mapping UvrD–elongation-complex interactions
We next mapped the location of UvrD within the elongation complex. Single-stranded/duplex DNA junctions are the preferred UvrD loading sites29; therefore, the upstream exposed portion of the transcription bubble30 is an optimal UvrD binding site. In this model, UvrD bound to RNAP pulls the elongation complex backwards while unwinding the upstream fork of the bubble (Fig. 3c). To locate UvrD within the elongation complex we incorporated photo-inducible 4[Author: 4′ (add prime symbol)?NO]-thio-deoxyuridine-5[Author: 5′ (add prime symbol)?YES]-monophosphate (sU[Author: is sU a standard nomenclature? NO what does the ‘s’ stand for?Thio; you can use 4-thio-U instead]) into the non-template strand at different positions (relative to catalytic site) to induce protein–DNA crosslinks. The −1 and −9 sU probes generated major [Author: can you rephrase major? Do you mean abundant or the most abundant? most abundant]crosslinked species corresponding to ββ′ subunits of RNAP; their pattern didn't change in the presence of UvrD (Fig. 4a, lanes 5–8). A unique UvrD-specific crosslinked species was detected only with the −9 probe (lane 6) and was consistent with the UvrD adduct. No significant crosslinks were detected in front of RNAP (position +22). These results support [Author: the model?YES]that UvrD binds near the upstream fork of the transcription bubble.
Figure 4. Mapping UvrD interactions with the elongation complex.

a, The 5′-radiolabelled scaffold carries a single photo-inducible 4-thio-dU (red) in the template strand (top). Protein crosslinking adducts corresponding to the β and β′ subunits of RNAoP and UvrD (red asterisk) (middle). A model of UvrD binding (bottom). b, UvrD (top, Protein Data Bank (PDB) accession number 2IS4)12 is cross-linked to RNAP (middle, PDB accession number 4IGC)47 at three positions (magenta) that span the β (yellow) and β′ (light blue) subunits, proximal to the non-template strand (blue, PDB accession number 4G7O). The β flap-tip-helix (green) is indicated and the suggested binding interface is circled. Schematic summarizing UvrD–RNAP binding shown below.
To further map interactions between UvrD and the elongation complex, we used bis[sulfosuccinimidyl] suberate (BS3) to crosslink proximal lysine residues between UvrD and RNAP. Using mass spectrometry we identified three inter-protein crosslinks between UvrD and RNAP. These crosslinks mapped to ββ′ subunits of RNAP and clustered around the DNA binding region on UvrD (Fig. 4b and Extended Data Fig. 6). In particular, one crosslink, (β K909)–(UvrD K124), mapped to the β flap tip domain. During elongation, the flap tip is required for activity of the NusA elongation factor31. We propose that UvrD also binds RNAP proximal to the flap tip domain in a way that allows UvrD to reach the non-template DNA strand near the upstream fork of the bubble. The positions of the remaining two crosslinks (β′ K79)–(UvrD K389) and (β′ K40)–(UvrD K448) are consistent with this hypothesis (Fig. 4b and Extended Data Fig. 6).
Considering the proximity of UvrD and NusA [Author: perhaps as the start of a new paragraph define what you are referring to by ‘their’?] on the surface of RNAP, and the fact that NusA potentiates RNAP backtracking32, we examined whether NusA supports UvrD-mediated backtracking. Indeed, NusA augmented UvrD-inducible arrests during elongation (Fig. 5a), providing a mechanistic explanation for the genetic evidence implicating NusA in Mfd-independent TCR14. Consistently, we showed that deletion of greB suppressed sensitivity of nusA cells (ΔnusA[Author: should this Δ and other Δ symbols be italic?NO] and nusA11) to 4NQO, mitomycin C, nitrofurazone (NFZ) and UV (Fig. 5b and Extended Data Fig. 7). It has been reported that UvrD and NusA directly bind UvrB and UvrA, respectively14,33,34. Thus, not only do the two elongation factors act synergistically to clear RNAP from the lesion site, they probably also recruit UvrAB to the damage site to facilitate repair (Fig. 5c).
Figure 5. UvrD and NusA cooperate in backtracking-mediated NER.
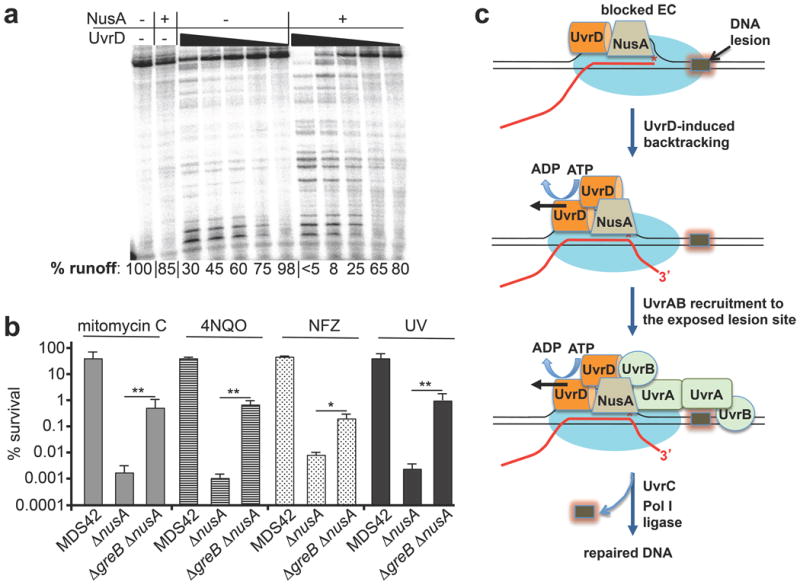
a, NusA facilitates UvrD-mediated backtracking. The fraction of full-length transcript (percentage runoff) is indicated. b, GreB inactivation in MDS43 cells suppresses ΔnusA sensitivity to mitomycin C (0.5 μg ml−1), 4NQO (3 μM), nitrofurazone (NFZ, 2 μM), and UV irradiation (15 J per m2). Data from three independent experiments are presented as the mean ± s.e.m.; *P < 0.05, **P < 0.01. c, Model for backtracking-mediated NER.
RNAP as a global DNA damage surveillance vehicle
A major challenge for NER is that UvrAB must recognize almost every type of bulky adduct on DNA among millions of normal base pairs in the genome. The aberrations detected by UvrAB range from apurinic/apyrimidinic (AP) sites to UV-induced photoproducts to large chemical adducts2. The accuracy and efficiency of the repair of such variable lesion types would be improved by a screening mechanism that recognizes a common structure or process, such as that of arrested RNAP during transcription elongation.
The preferential repair of the template DNA strand, that is, TCR, implies that the transcription apparatus augments the search and/or binding of lesions by NER enzymes3. Cellular RNAPs are highly sensitive to aberrations in the template strand, and are temporally or permanently blocked by various DNA adducts22–24,35. As bacterial and eukaryotic genomes are pervasively transcribed, RNAP can serve as a global surveyor of DNA damage that constantly monitors the quality of DNA via its natural one-dimensional diffusion. However, when stopped at DNA lesions, RNAP must either be terminated or moved aside to maintain damage site accessibility for NER. The Mfd pathway of TCR necessitates transcription termination10, whereas the UvrD pathway described here uses RNAP backtracking. Importantly, this new UvrD mechanism facilitates NER without loss of RNAP, enabling transcription to promptly resume after DNA repair.
Mfd-independent TCR
The present work argues that the bulk of TCR occurs via UvrD-dependent backtracking (Fig. 5c); the sensitivity of uvrD and nusA cells to UV and various DNA damaging agents is much greater than that of mfd cells (Figs 3, 5b and Extended Data Figs 4, 7, 8). Yet, promoting backtracking by eliminating Gre factors, or by slowing ribosomal translocation, eliminates much of the DNA damage-induced lethality associated with UvrD or NusA deficiency (Figs 3, 5b and Extended Data Figs 4, 7). Consistently, deletion of DksA, a factor that competes with Gre for the secondary channel of RNAP, also increases E. coli sensitivity to mitomycin C36. Remarkably, deletion of mfd itself prominently suppresses uvrD sensitivity to UV and mitomycin C (Fig. 3b and Extended Data Figs 4, 8). This is consistent with the anti-backtracking mechanism of Mfd8, further supporting the notion that RNAP backtracking is a prerequisite for the majority of [Author: can ‘the majority of’ be changed to ‘most’?OK]NER events (Fig. 3d). Survival after genotoxic challenge depends on both efficient NER and swift recovery from the SOS response. Indeed, greAB deletions facilitate NER, but do not increase cell survival (Fig. 3a, b). Similarly, Mfd function may not be as important for TCR per se, but instead for cellular recovery after the excessive backtracking associated with the SOS response.
In the absence of cellular stress, UvrD is equimolar with RNAP (∼3,000 molecules per cell37). During the SOS response, however, the intracellular level of UvrD increases approximately threefold11. This spike facilitates UvrD dimerization and helicase activity38 and probably is a prerequisite for UvrD-mediated backtracking. A sigmoid-shaped graph of RNAP arrest as a function of UvrD concentration (Extended Data Fig. 9) supports this view. After washing, UvrD remains bound to RNAP (Extended Data Fig. 1b), yet loses backtracking ability (Fig. 1a), suggesting monomeric and multimeric associations with RNAP during periods of no stress and stress, respectively (Fig. 5c). Excessive backtracking can be detrimental to genomic integrity in cells recovering from genotoxic stress and resuming replication, as frequent co-directional collisions between the replisome and backtracked elongation complexes result in dsDNA breaks (DSBs)39. By ‘pushing’ backtracked RNAPs forward, Mfd suppresses DSBs associated with such collisions39, and hence diminishes the high frequency of mutations associated with DSBs repair. This model is consistent with the reduced ‘mutation frequency decline’ phenotype of mfd cells as well as their high UV mutability, minimal UV sensitivity40, and compromised transcriptional recovery after UV exposure41.
The process of TCR and the basic structural organization of RNAPs are evolutionarily preserved1,6,26. Bulky DNA lesions, such as thymine dimers, stall bacterial and eukaryotic RNAPs similarly22,23,42. As backtracking is a fundamental feature of all RNAPs17 and occurs pervasively throughout the eukaryotic genome43, it is likely to drive TCR in higher organisms as well. Notably, mammalian [Author: human?]3′-5′ DNA helicase, XPB, and its homologs from other eukaryotes, associates with elongating RNAP II as a subunit of the TFIIH transcription factor. It has been implicated in NER as well as in several human disorders associated with deficient DNA repair44,45. It is thus possible that the mechanistic role of XPB is analogous to that described here for UvrD, that is, backtrack-inducing TCR.
Methods Summary
Cultures of E. coli were crosslinked with formaldehyde, collected by centrifugation, and lysed. RNAP-containing material was pulled down with anti-RNAP antibodies and proteins were identified by mass spectrometry. For DNA damage experiments, colony-forming units were counted after exposure to increasing concentrations of 4-NQO, mitomycin C or cisplatin. For UV survival assays, diluted cultures were plated, irradiated with a UV lamp, and incubated overnight in the dark. Strains were transformed with pUC18 plasmid and induced with IPTG before and after irradiation. In vitro transcription assays were performed as described16. RNA-DNA scaffolds containing cyclo-thymine dimers or 4-thio-dUMP-modified template oligonucleotides were assembled with immobilized, biotinylated RNAP before incubation with components of UvrABC system or irradiation at 308 nM for protein–DNA crosslinking, respectively. Purified RNAP and UvrD were crosslinked with BS3, and crosslinked proteins were reduced with dithiothreitol (DTT), alkylated with iodoacetamide and digested with trypsin. Tryptic peptides were analysed with a mass spectrometer coupled to a liquid chromatography system. pLink46 was used to search Mascot generic format files for inter-peptide crosslinks. For in situ DNA footprinting, the uvrD gene was cloned into a vector under the PLtetO-1 promoter, and induced with anhydrotetracycline for 1 h before CAA modification. CAA modifications on non-template DNA were analysed with primer extension using [32P]-labelled primer in parallel with the sequencing reactions.
Methods
Bacterial strains
The strains used in this study are listed in Extended Data Table 1 and were constructed using standard genetic techniques.
Mass spectrometry of in vivo RNAP interactome
Mid-exponential cultures of E. coli K12 MG1655 were treated with formaldehyde (Thermo Fisher, final concentration 50 mM) for 30 min, cells were collected by centrifugation, and lysed by a combination of ultrasound disruption and the action of lysozyme (human recombinant, Sigma-Aldrich). Pull-down of RNAP-containing material was performed using a mix of anti-RNAP antibodies (Neoclone) immobilized on Protein A/G Mag Sepharose beads (GE). Protein identification in the pull-down material was carried out at the NYUMC Protein Core Mass Spectrometry Facility, using an LTQ-Orbitrap mass spectrometer followed by Mascot database search (Matrixscience). Protein abundance was estimated using emPAI scores15. The range of emPAI values for a representative set of proteins in RNAP pull-downs spans 3 independent experiments (Extended Data Fig. 1).
Measuring sensitivity to DNA damage
E. coli strains were cultured in LB medium overnight. Appropriate dilutions were spread on LB agar plates containing increasing concentrations of 4-NQO or NFZ or mitomycin C or cisplatin, followed by an overnight incubation at permissive temperatures. Colony-forming units (c.f.u.) were counted at each concentration of genotoxic agents. Chloramphenicol was used at the sublethal concentration (1.0 μg ml−1), along with genotoxic agents. A 10 mM stock solution of NFZ or 4-NQO was first made in N,N-dimethylformamide, stored at −20 °C, and diluted appropriately for each experiment. Mitomycin C was dissolved in sterile saline to a concentration of 500 μg ml−1 and stored at −20 °C. Cisplatin was dissolved in LB medium to a concentration of 500 μg ml−1 just before use. For UV survival assays, overnight grown cultures were diluted in M9 minimal salts, appropriate dilutions were spread on LB agar plates and irradiated with a UV (254 nm) lamp and incubated overnight in the dark at appropriate temperatures.
In vivo TCR
Strains MG1655Z1 and MG1655Z1ΔgreAΔgreB were transformed with pUC18 plasmid. Overnight cultures were diluted 1:100 in LB medium supplemented with 100 μg ml−1 ampicillin and grown until OD600 ∼0.3 at 30 °C. The culture was centrifuged at 5000 rpm for 5 min. The pellet was resuspended in M56 to an absorbance of 0.4.
The resuspended culture was spread on a Petri dish and, whilst being shaken, irradiated with 70 J per m2 of 254 nm UV light. To induce the transcription on pUC18, 1 mM isopropyl-β-d-thiogalactoside (IPTG) was added to the culture medium 1 h before and immediately after UV irradiation. The irradiated culture was then supplemented with 0.25% casamino acids, 0.004% thiamine and 0.4% glucose and incubated at 30 °C in the dark to avoid removal of cyclobutane dimers by photoreactivation. Cells were collected before UV irradiation (-), immediately after (time 0 repair) and at defined repair times (15, 30, 45 and 60 min). Plasmid DNA was isolated from the cultures using a Qiagen plasmid isolation kit.
Primer extension was performed with (32P)-labelled primers for analysis of cyclobutane pyrimidine dimers on the template strand. End labelled primer, 50 pmol, (5′-GGCATGCAAGCTTGGCACTGGC-3′), 400 μM dNTPs, 200 ng of plasmid DNA and Taq DNA polymerase were mixed in Thermopol buffer. The mixture was denatured by heating to 98 °C in a thermal cycler for 5 min followed by annealing at 55 °C for 5 min. Primer extension was performed at 68 °C for 6 min. Formamide stop solution was added to the reaction mixture and aliquots loaded onto a 6% urea-polyacrylamide gel. Percentages of repaired DNA were calculated using Image-Quant (GE) and averaged from three independent experiments with untreated samples taken as 100%.
In vitro transcription assays
E. coli RNAP and UvrD were purified as described in refs 16 and 48, respectively. DNA templates were constructed using the non-transcribed part of T7 A1 promoter fragments fused to appropriate transcription units. Resulting DNA fragments were PCR amplified and purified from an agarose gel. For biotinylated DNA templates, biotinylated DNA oligonucleotides (IDT) were used. All NTP substrates were purified by ion-exchange chromatography16. Transcription at the T7A1-promoter templates was initiated by mixing 1 pmol of RNAP with equimolar amount of appropriate DNA PCR fragments in TB50 buffer (40 mM Tris-HCl pH 8.0; 10 mM MgCl2; 50 mM NaCl) followed by addition of 10 μM ApUpC RNA primer, 25 μM ATP and GTP. Incubation was continued for 5 min at 37 °C. Resulting complexes were labelled by addition of 2 μCu α-[32P]-CTP (3,000 Ci mmol−1; MP Biomedicals) for 5 min at room temperature and immobilized on Neutravidin beads (5–10 μl; Piers) in the presence of 1.5 mg ml−1 heparin. The resulting EC20 was washed twice with 1 ml of TB1000 (TB with 1 M NaCl), twice with 1 ml of TB100 and divided into 10 μl aliquots. For the chase experiments, the EC20 was incubated with either the indicated amounts of UvrD or equal volumes of mock buffer for 5 min at room temperature and then chased for 5 min at 37 °C by addition of 0.1 mM NTPs. Where indicated, beads were washed after the chase with 1 ml of TB1000 and 1 ml of TB100. All reactions were stopped by equal volume of Stop Buffer (SB, 1×TBE buffer, 8 M Urea, 20 mM EDTA, 0.025% xylene cyanol, 0.025% bromophenol blue). For the GreB cleavage assay (Fig. 2a), two 10 μl aliquots were incubated for 5 min at room temperature with 1 μM GreB. One aliquot was then chased for 5 min by addition of 0.1 mM NTPs and both samples were quenched as above. To test the effect of NusA on UvrD-induced backtracking (Fig. 5a), the initial EC20 was prepared as above and incubated with the indicated amounts of UvrD in the absence or presence of 10 nM NusA. Reaction mixtures were chased as above. For the UvrD-induced formation of stalled ECs (Fig. 2b), EC20 was walked to position 36–39 as described in ref. 16. The resulting ECs were mixed with 12.5 nM UvrD and 1 mM ATP or an equal amount of mock solution and incubated at 37 °C for the indicated time intervals. Next, 10 μl aliquots were withdrawn and mixed with 0.1 mM GTP and CTP for 5 min at room temperature. Reactions were stopped by addition of equal volumes of SB. The amount of UvrABC-mediated DNA cleavage products after 30 min at 37 °C was taken as 100% and UvrABC-mediated cleavage after CTP + GTP chase was calculated as a fraction of that value. Image-Quant (GE) software was used for all quantifications.
Reconstitution of the EC and UvrABC-mediated processing of thymine dimers
For assembly of the RNA-DNA scaffold (Fig. 2a), 75 pmol A1 RNA (IDT) were mixed with 75 pmol of the cyclo-thymine dimer template DNA strand or control strand (Midland, TX) in 20 μl of the annealing buffer (AB) (12% glycerol; 20 mM Tris pH 8.0; 40 mM KCl; 5 mM MgCl2) and annealed gradually in a PCR cycler. The resulting scaffold was stored at −20°C and used as needed. To measure the extent of UvrABC-directed cleavage, 15 pmol of the DNA–RNA scaffold were mixed with 15 pmol of biotinylated RNAP in 20 μl of AB and incubated for 10 min at room temperature. 75 pmol of non-template strand was added and incubation continued for another 5 min. The resulting complex was immobilized at Neutravidin-coated beads (Pierce), washed twice with TB1000 and then with TB100. For RNA labelling, 5 μM ATP and 2 μCu α-[32P]-CTP were added for 5 min at room temperature, followed by washing with TB100. For the UvrC cleavage reaction complexes were washed twice with TB0 (20 mM Tris-HCl pH 8.0; 10 mM MgCl2) and 10× PNK buffer (New England Biolabs) was added together with 1 μCu γ-[32P]-ATP (7,000 Ci mmol−1; MP Biomedicals) and 20 units PNK (New England Biolabs). The DNA labelling reaction continued for 30 min at 37 °C, then the beads (EC11) were washed twice with TB1000 and TB100. EC11 were processed directly or chased first for 10 min at 37 °C by the addition of 1 mM NTPs. Complexes were supplemented with 1 mM ATP and 12.5 nM UvrD was added where indicated for 10 min at 37 °C before washing with TB100. To initiate the cleavage reaction premixed UvrA (2.5 nM), UvrB (10 nM) and UvrC (25 nM) were added and incubated for the indicated time intervals at 37 °C before quenching with SB. UvrABC were purified as described in ref. 49. Products of all reactions were separated in 6–23% denaturing polyacrylamide gels and visualized via phosphor-imaging using a Typhoon phosphor-imager (GE Healthcare) and Image Quant software (GE Healthcare).
Protein–DNA crosslinking
RNA–DNA scaffolds were assembled as above using the 4-thio-dUMP-modified template oligos (Midland, TX), A1 RNA (9 nt long), and non-template DNA. Resulting complexes were labelled as described above and incubated with 12.5 nM UvrD or mock solution for 5 min at 37 °C before being irradiated for 10 min with a hand-held UV lamp (Cole Parmer) at 308 nM wavelength. Irradiation was performed on ice and samples were denatured in Laemmli loading buffer. Products were separated in a 6% denaturing polyacrylamide gel with 0.1% SDS and Tris-glycine running buffer. UvrD adducts were seen on the gel as a 90 kDa species.
UvrD-RNAP binding assay
UvrD and RNAP, UvrD alone, and RNAP alone were incubated at room temperature for 30 min in binding buffer (50 mM Tris pH 8.0, 100 mM NaCl, 50 mM imidazole) before they were added to His Mag Sepharose Ni beads (GE), which were pre-equilibrated and washed with binding buffer. Each was then incubated for 30 min with gentle agitation at room temperature. After incubation, beads were washed three times with binding buffer and eluted in 50 μl of elution buffer (50 mM Tris pH 8.0, 100 mM NaCl, 400 mM imidazole). Samples were electrophoresed in an SDS-polyacrylamide gel alongside pure UvrD, for reference.
RNAP-UvrD cross-linking and their mapping by LC-MS/MS
Purified RNAP at a final concentration of 2 μM was incubated with 4 μM UvrD in crosslinking buffer (1× PBS, 500 mM NaCl, pH 7.4) for 30 min at room temperature. Crosslinking was initiated upon addition of Bis(sulfosuccinimidyl) suberate (BS3) (Thermo Scientific) in 1×PBS at a final concentration of 350 μM. Crosslinking reactions continued for 30 min at room temperature before they were quenched with 1 M Tris pH 8.0 at a final concentration of 200 mM.
Crosslinked proteins were separated by SDS–PAGE (4–12% Bis Tris Novex gels, Invitrogen) and stained with GelCode Blue. Super-shifted (relative to positions of untreated RNAP subunits) were removed from the gel and destained. Samples were reduced with 20 mM DTT (Sigma) at 57 °C for 60 min and alkylated with 45 mM iodoacetamide (Sigma) in the dark for 45 min at room temperature before overnight, in-gel digestion with 0.5 μg sequencing grade modified trypsin (Promega).
Gel slices were incubated with Poros R2 50 μm slurry diluted in 5% formic acid and 0.2% TFA to bind tryptic peptides. The Poros bead slurry was collected and washed through C18 Zip Tips (Millipore) before elution with 40 μl of 40% acetonitrile in 0.5% acetic acid. Eluted peptides were dehydrated in vacuum and resuspended in 40 μl 0.5% acetic acid for MS analysis.
Peptide aliquots (∼200 ng) were analysed in the Q Exactive mass spectrometer (Thermo Scientific) coupled to an EASY-nLC (Thermo Scientific) liquid chromatography system. The peptides were eluted over a 120-min linear gradient from 98% buffer A (water) to 40% buffer B (ACN) then continue to 100% buffer B over 10 min with a flow range of 200 nl min−1. Each full MS scan (R = 70,000) was followed by dd-MS2 (R = 17,500) with HCD and an isolation window of 2.0 m/z. Normalized collision energy was set to 35. Precursors of +1, +2 and +3 were excluded from MS2 scans; monoisotopic screening was enabled and a dynamic exclusion window was set to 30.0 s.
Identification of cross-links: Mascot generic format (mgf) files were generated from Xcalibur raw files using the Proteome Discoverer software (Thermo Scientific). pLink46 was used to search resulting mgf files for interpeptide crosslinks with the following settings: precusor mass tolerance 20 p.p.m.; fragment mass tolerance 5 p.p.m.; BS3 crosslinker (crosslink monoisotopic mass shift of 138.0680786 Da, monolink mass shift of 156.0786442 Da. One fixed modification (Carbamidomethyl-C) and three variable modifications (oxidation-M, N-terminal glutamate to pyroglutamate, N-terminal acetylation) were specified. False discovery rate was set at <5%. Fragmentation spectra for each crosslinked peptide were validated using pLabel50 and confirmed with manual inspection.
In situ DNA footprinting
To construct the plasmid pUvrD (Cmr, p15a), the uvrD gene was excised from the plasmid pETDuet-uvrD as an XhoI / filled-in XbaI fragment and cloned into the pZA31 vector at the SalI site (compatible cohesive end with XhoI) and the blunted KpnI site. The expression of the uvrD gene in the resulting plasmid is driven by the PL-derivative promoter PLtetO-1 which is controlled by the operator repressor system of the Tn10-derived tet resistance operon51. The induction of PLtetO-1 is achieved by anhydrotetracycline.
To ensure expression of the regulatory repressor proteins LacI and TetR, the entire unit encoding LacR, TetR and Spr53 was transferred into the chromosome of E. coli strain MG1655 by P1 transduction (MG1655Z1).
E. coli strains were grown in 10 ml of M9 medium supplemented with 0.4% glucose, 4 mg ml−1 casamino acids, 100 μg ml−1 ampicillin and 30 μg ml−1 chloramphenicol. To induce the PLtetO-1 promoter, anhydrotetracycline (50 ng ml−1) was added for 1 h before CAA modification. At OD600∼0.6 1 mM IPTG was added (if required) for 10 min, then CAA was added to 4% final concentration and the incubation was continued for 7 min. Then cells were collected, immediately washed with the saline solution, collected and the plasmid DNA extracted.
To analyse CAA modification52 of the non-template DNA strand, primer extension was performed with [32P]-labelled primer (5′-TAGCTTCCTTAGCTCCTGA-3′) in parallel with the sequencing reactions in a cycler at the following conditions: 94 °C for 3 min (initial denaturation), then 94 °C for 30 s (denaturation), 56 °C for 30 s (annealing), 72 °C for 60 s (extension), 25 cycles total. Formamide stop solution was added to the reaction mixture and aliquots were analysed by electrophoresis in an 8% urea-polyacrylamide gel.
Extended Data
Extended Data Figure 1. UvrD binds RNAP.

a, A sample list of RNAP-bound proteins from exponentially grown E. coli. Abundance is based on emPAI score15. b, UvrD + 6His–RNAP, UvrD, or 6His–RNAP were affinity-purified on nickel beads and electrophoresed alongside pure UvrD, for reference; The asterisk indicates UvrD. c, RNAP-UvrD complex isolated by size-exclusion chromatography. UvrD–RNAP were mixed 3:1, and run over a Superdex 200 10/300GL column. RNAP core (red) and UvrD (green) chromatograms are overlaid for comparison. The inlaid polyacrylamide gel displays fractions taken from each chromatographic peak.
Extended Data Figure 2. UvrD promotes long-range backtracking.

Biotinylated RNAP was used to prepare the startup EC11 immobilizing on beads. It was walked to position 39, followed by incubation with UvrD, washing (to remove UvrD and NTPs), and then treatment with GreB for indicated times. Numbers on the right indicate the size of 5′-labelled RNAs.
Extended Data Figure 3. Thymine dimer in the template strand blocks the elongation complex.

Schematic diagram of the T7A1 promoter template shows the position of CPD (red). Both control (no TT) and CPD-bearing template strands were radiolabelled at their 5′ ends (top band). The sequencing gel also shows the radiolabelled RNA products before and after chase of EC11. The EC was completely halted by CPD (indicated by red TT). On the control template it formed the runoff products.
Extended Data Figure 4. Effect of greAB, mfd and ribosome inactivation on uvrD sensitivity to DNA damaging agents and UV.

a, Representative efficiencies of colony formation of wild-type (MG1655) and mutant E. coli cells in the presence of the indicated amounts of mitomycin C, 4NQO and cisplatin. Cells were spotted on LB agar plates in serial tenfold dilutions and incubated at 30 °C for 24 h. b, Representative efficiencies of colony formation of wild-type and ΔuvrD cells in the presence of the indicated amounts of mitomycin, 4NQO and chloramphenicol. Cells were spotted on LB agar plates in serial tenfold dilutions and incubated at 30 °C for 24 h. c, Representative efficiencies of colony formation of wild-type (MG1655) and mutant E. coli cells after UV irradiation. Cells were spotted on LB agar plates in serial tenfold dilutions and incubated at 30 °C for 24 h. d, Representative efficiencies of colony formation of wild-type and mutant E. coli cells after UV irradiation in the presence of a sublethal dose of chloramphenicol. Cells were spotted on LB agar plates in serial tenfold dilutions and incubated at 30 °C for 24 h.
Extended Data Figure 5. UvrD inactivation suppresses temperature sensitivity of greAB cells.
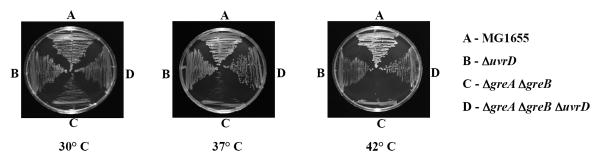
E. coli strains were streaked on LB agar plates and incubated at the indicated temperatures for 24 h.
Extended Data Figure 6. UvrD–RNAP crosslinks.

a, Three inter-protein crosslinks indicate that UvrD binds near the β flap on RNAP. UvrD (grey, PDB accession code 2IS4) cross-links to RNAP (PDB accession code 4IGC) at three distinct positions that span the β (pale yellow) and β′ (light blue) subunits of RNAP. The non-template strand (blue, PDB accession code 4G7O) is indicated for reference. Crosslinked lysines are colored magenta and pairs are connected with a black line. b, MS2 spectrum of a representative crosslinked pair (β′ K40)–(UvrD K448). The peptide sequences with cross-linked lysine residues are shown (top right). Observed peptide backbone cleavage is indicated and b- and y-type fragment ions are labelled in the spectrum. The m/z tolerances of fragment ions are presented in the inset below the spectra. Spectra were annotated using pLabel50.
Extended Data Figure 7. GreB inactivation suppresses nusA sensitivity to genotoxic chemicals and UV.

a, Representative efficiencies of colony formation of wild-type (MG1655) and mutant E. coli cells in the presence of indicated amounts of NFZ and 4NQO. Cells were spotted on LB agar plates in serial tenfold dilutions and incubated at 30 °C for 24 h. b, Data from three independent experiments are presented as the mean ± s.e.m.; **P < 0.01. c, Representative efficiencies of colony formation of MDS42 and mutant E. coli cells in the presence of indicated amounts of mitomycin, 4NQO, NFZ and after UV irradiation. Cells were spotted on LB agar plates in serial tenfold dilutions and incubated at 30 °C for 24 h.
Extended Data Figure 8. Deletion of mfd partially suppresses uvrD sensitivity to mitomycin C.

a, Representative efficiencies of colony formation of wild-type (MG1655) and mutant E. coli cells in the presence of indicated amounts of mitomycin C. Cells were spotted on LB agar plates in serial tenfold dilutions and incubated at 30 °C for 24 h. b, Data from three independent experiments are presented as the mean ±s.e.m.; **P < 0.01.
Extended Data Figure 9. Transcriptional arrest as a function of UvrD concentration.

a, A representative chase experiment demonstrating multiple transcriptional arrests as a function of UvrD concentration. b, Data from three independent experiments are plotted as the mean ± s.e.m. Arrest efficiency (%) was calculated as a fraction of all arrested complexes in relation to the full-length runoff.
Extended Data Table 1 Escherichia coli strains used in this study.
Acknowledgments
We thank D. Jeruzalmi for materials. This work was supported by the Russian Foundation for Basic Research (A.M.) and the NIH, BGRF, Dynasty foundation and by the Howard Hughes Medical Institute (E.N.).
Footnotes
Author Contributions: V.E., V.K., K.M., V.S., B.U., S.P. and A.M. conducted the experimental work, discussed the results and commented on the manuscript. E.N. designed the study and wrote the paper.
The authors declare no competing financial interests. Readers are welcome to comment on the online version of the paper.
References
- 1.Reardon JT, Sancar A. Nucleotide excision repair. Prog Nucleic Acid Res Mol Biol. 2005;79:183–235. doi: 10.1016/S0079-6603(04)79004-2. Medline CrossRef. [DOI] [PubMed] [Google Scholar]
- 2.Van Houxen B, McCullough A. Nucleotide excision repair in E. coli. Ann NY Acad Sci. 1994;726:236–251. doi: 10.1111/j.1749-6632.1994.tb52822.x. Medline CrossRef. [DOI] [PubMed] [Google Scholar]
- 3.Ganesan A, Spivak G, Hanawalt PC. Transcription-coupled DNA repair in prokaryotes. Prog Mol Biol Transl Sci. 2012;110:25–40. doi: 10.1016/B978-0-12-387665-2.00002-X. Medline CrossRef. [DOI] [PubMed] [Google Scholar]
- 4.Truglio JJ, Croteau DL, Van Houten B, Kisker C. Prokaryotic nucleotide excision repair: the UvrABC system. Chem Rev. 2006;106:233–252. doi: 10.1021/cr040471u. Medline CrossRef. [DOI] [PubMed] [Google Scholar]
- 5.Mellon I, Hanawalt PC. Induction of the Escherichia coli lactose operon selectively increases repair of its transcribed DNA strand. Nature. 1989;342:95–98. doi: 10.1038/342095a0. Medline CrossRef. [DOI] [PubMed] [Google Scholar]
- 6.Gaillard H, Aguilera A. Transcription coupled repair at the interface between transcription elongation and mRNP biogenesis. Biochim Biophys Acta. 2013;1829:141–150. doi: 10.1016/j.bbagrm.2012.09.008. Medline CrossRef. [DOI] [PubMed] [Google Scholar]
- 7.Deaconescu AM, et al. Structural basis for bacterial transcription-coupled DNA repair. Cell. 2006;124:507–520. doi: 10.1016/j.cell.2005.11.045. Medline CrossRef. [DOI] [PubMed] [Google Scholar]
- 8.Park JS, Marr MT, Roberts JW. E. coli Transcription repair coupling factor (Mfd protein) rescues arrested complexes by promoting forward translocation. Cell. 2002;109:757–767. doi: 10.1016/s0092-8674(02)00769-9. Medline CrossRef. [DOI] [PubMed] [Google Scholar]
- 9.Savery N. Prioritizing the repair of DNA damage that is encountered by RNA polymerase. Transcription. 2011;2:168–172. doi: 10.4161/trns.2.4.16146. Medline CrossRef. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Selby CP, Sancar A. Molecular mechanism of transcription-repair coupling. Science. 1993;260:53–58. doi: 10.1126/science.8465200. Medline CrossRef. [DOI] [PubMed] [Google Scholar]
- 11.Kumura K, Sekiguchi M. Identification of the uvrD gene product of Escherichia coli as DNA helicase II and its induction by DNA-damaging agents. J Biol Chem. 1984;259:1560–1565. Medline. [PubMed] [Google Scholar]
- 12.Lee JY, Yang W. UvrD helicase unwinds DNA one base pair at a time by a two-part power stroke. Cell. 2006;127:1349–1360. doi: 10.1016/j.cell.2006.10.049. Medline CrossRef. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matson SW, George JW. DNA of helicase II Escherichia coli. Characterization of the single-stranded DNA-dependent NTPase and helicase activities. J Biol Chem. 1987;262:2066–2076. Medline. [PubMed] [Google Scholar]
- 14.Cohen SE, et al. Roles for the transcription elongation factor NusA in both DNA repair and damage tolerance pathways in Escherichia coli. Proc Natl Acad Sci USA. 2010;107:15517–15522. doi: 10.1073/pnas.1005203107. Medline CrossRef. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ishihama Y, et al. Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol Cell Proteomics. 2005;4:1265–1272. doi: 10.1074/mcp.M500061-MCP200. Medline CrossRef. [DOI] [PubMed] [Google Scholar]
- 16.Nudler E, Gusarov I, Bar-Nahum G. Methods of walking with the RNA polymerase. Methods Enzymol. 2003;371:160–169. doi: 10.1016/S0076-6879(03)71011-8. Medline CrossRef. [DOI] [PubMed] [Google Scholar]
- 17.Nudler E. RNA polymerase backtracking in gene regulation and genome instability. Cell. 2012;149:1438–1445. doi: 10.1016/j.cell.2012.06.003. Medline CrossRef. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Borukhov S, Sagitov V, Goldfarb A. Transcript cleavage factors from E. coli. Cell. 1993;72:459–466. doi: 10.1016/0092-8674(93)90121-6. Medline CrossRef. [DOI] [PubMed] [Google Scholar]
- 19.Nudler E, Mustaev A, Lukhtanov E, Goldfarb A. The RNA-DNA hybrid maintains the register of transcription by preventing backtracking of RNA polymerase. Cell. 1997;89:33–41. doi: 10.1016/s0092-8674(00)80180-4. Medline CrossRef. [DOI] [PubMed] [Google Scholar]
- 20.Brosh RM, Jr, Matson SW. Mutations in motif II of Escherichia coli DNA helicase II render the enzyme nonfunctional in both mismatch repair and excision repair with differential effects on the unwinding reaction. J Bacteriol. 1995;177:5612–5621. doi: 10.1128/jb.177.19.5612-5621.1995. Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Proshkin S, Rahmouni AR, Mironov A, Nudler E. Cooperation between translating ribosomes and RNA polymerase in transcription elongation. Science. 2010;328:504–508. doi: 10.1126/science.1184939. Medline CrossRef. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Donahue BA, Yin S, Taylor JS, Reines D, Hanawalt PC. Transcript cleavage by RNA polymerase II arrested by a cyclobutane pyrimidine dimer in the DNA template. Proc Natl Acad Sci USA. 1994;91:8502–8506. doi: 10.1073/pnas.91.18.8502. Medline CrossRef. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Selby CP, Drapkin R, Reinberg D, Sancar A. RNA polymerase II stalled at a thymine dimer: footprint and effect on excision repair. Nucleic Acids Res. 1997;25:787–793. doi: 10.1093/nar/25.4.787. Medline CrossRef. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Selby CP, Sancar A. Transcription preferentially inhibits nucleotide excision repair of the template DNA strand in vitro. J Biol Chem. 1990;265:21330–21336. Medline. [PubMed] [Google Scholar]
- 25.Manelyte L, Kim YI, Smith AJ, Smith RM, Savery NJ. rate enhancement during transcription-coupled DNA repair. Mol Cell. 2010;40:714–724. doi: 10.1016/j.molcel.2010.11.012. Medline CrossRef. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Batty DP, Wood RD. Damage recognition in nucleotide excision repair of DNA. Gene. 2000;241:193–204. doi: 10.1016/s0378-1119(99)00489-8. Medline CrossRef. [DOI] [PubMed] [Google Scholar]
- 27.Doudney CO, Rinaldi CN. Chloramphenicol-promoted increase in resistance to UV damage in Escherichia coli B/r WP2 trpE65: development of the capacity for successful repair of otherwise mutagenic or lethal lesions in DNA. Mutat Res. 1985;143:29–34. doi: 10.1016/0165-7992(85)90100-9. Medline CrossRef. [DOI] [PubMed] [Google Scholar]
- 28.Hanawalt PC. The U.V. sensitivity of bacteria: its relation to the DNA replication cycle. Photochem Photobiol. 1966;5:1–12. Medline CrossRef. [PubMed] [Google Scholar]
- 29.Tomko EJ, et al. 5′-Single-stranded/duplex DNA junctions are loading sites for E. coli UvrD translocase. EMBO J. 2010;29:3826–3839. doi: 10.1038/emboj.2010.242. Medline CrossRef. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Korzheva N, et al. A structural model of transcription elongation. Science. 2000;289:619–625. doi: 10.1126/science.289.5479.619. Medline CrossRef. [DOI] [PubMed] [Google Scholar]
- 31.Toulokhonov I, Artsimovitch I, Landick R. Allosteric control of RNA polymerase by a site that contacts nascent RNA hairpins. Science. 2001;292:730–733. doi: 10.1126/science.1057738. Medline CrossRef. [DOI] [PubMed] [Google Scholar]
- 32.Bar-Nahum G, et al. A ratchet mechanism of transcription elongation and its control. Cell. 2005;120:183–193. doi: 10.1016/j.cell.2004.11.045. Medline CrossRef. [DOI] [PubMed] [Google Scholar]
- 33.Ahn BA. physical interaction of UvrD with nucleotide excision repair protein UvrB. Mol Cells. 2000;10:592–597. doi: 10.1007/s10059-000-0592-5. Medline CrossRef. [DOI] [PubMed] [Google Scholar]
- 34.Manelyte L, et al. The unstructured C-terminal extension of UvrD interacts with UvrB, but is dispensable for nucleotide excision repair. DNA Repair (Amst) 2009;8:1300–1310. doi: 10.1016/j.dnarep.2009.08.005. Medline CrossRef. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tornaletti S. Transcription arrest at DNA damage sites. Mutat Res. 2005;577:131–145. doi: 10.1016/j.mrfmmm.2005.03.014. Medline CrossRef. [DOI] [PubMed] [Google Scholar]
- 36.Trautinger BW, Jaktaji RP, Rusakova E, Lloyd RG. RNA polymerase modulators and DNA repair activities resolve conflicts between DNA replication and transcription. Mol Cell. 2005;19:247–258. doi: 10.1016/j.molcel.2005.06.004. Medline CrossRef. [DOI] [PubMed] [Google Scholar]
- 37.Arthur HM, Eastlake PB. Transcriptional control of the uvrD gene of Escherichia coli. Gene. 1983;25:309–316. doi: 10.1016/0378-1119(83)90235-4. Medline CrossRef. [DOI] [PubMed] [Google Scholar]
- 38.Maluf NK, Fischer CJ, Lohman TM. A dimer of Escherichia coli UvrD is the active form of the helicase in vitro. J Mol Biol. 2003;325:913–935. doi: 10.1016/s0022-2836(02)01277-9. Medline CrossRef. [DOI] [PubMed] [Google Scholar]
- 39.Dutta D, Shatalin K, Epshtein V, Gottesman ME, Nudler E. Linking RNA polymerase backtracking to genome instability in E coli. Cell. 2011;146:533–543. doi: 10.1016/j.cell.2011.07.034. Medline CrossRef. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Witkin EM. Mutation and the repair of radiation damage in bacteria. Radiat Res. 1966;6(suppl):30–53. Medline CrossRef. [PubMed] [Google Scholar]
- 41.Schalow BJ, Courcelle CT, Courcelle J. Mfd is required for rapid recovery of transcription following UV-induced DNA damage but not oxidative DNA damage in Escherichia coli. J Bacteriol. 2012;194:2637–2645. doi: 10.1128/JB.06725-11. Medline CrossRef. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brueckner F, Cramer P. DNA photodamage recognition by RNA polymerase II. FEBS Lett. 2007;581:2757–2760. doi: 10.1016/j.febslet.2007.05.014. Medline CrossRef. [DOI] [PubMed] [Google Scholar]
- 43.Churchman LS, Weissman JS. Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature. 2011;469:368–373. doi: 10.1038/nature09652. Medline CrossRef. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Compe E, Egly JM. TFIIH: when transcription met DNA repair. Nature Rev Mol Cell Biol. 2012;13:343–354. doi: 10.1038/nrm3350. Medline CrossRef. [DOI] [PubMed] [Google Scholar]
- 45.Drapkin R, et al. Dual role of TFIIH in DNA excision repair and in transcription by RNA polymerase II. Nature. 1994;368:769–772. doi: 10.1038/368769a0. Medline CrossRef. [DOI] [PubMed] [Google Scholar]
- 46.Yang B, et al. Identification of cross-linked peptides from complex samples. Nature Methods. 2012;9:904–906. doi: 10.1038/nmeth.2099. Medline CrossRef. [DOI] [PubMed] [Google Scholar]
- 47.Murakami KS. X-ray crystal structure of Escherichia coli RNA polymerase σ70 holoenzyme. J Biol Chem. 2013;288:9126–9134. doi: 10.1074/jbc.M112.430900. Medline CrossRef. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Runyon GT, Wong I, Lohman TM. Overexpression, purification, DNA binding, and dimerization of the Escherichia coli uvrD gene product (helicase II) Biochemistry. 1993;32:602–612. doi: 10.1021/bi00053a028. Medline CrossRef. [DOI] [PubMed] [Google Scholar]
- 49.Pakotiprapha D, Samuels M, Shen K, Hu JH, Jeruzalmi D. Structure and mechanism of the UvrA-UvrB DNA damage sensor. Nature Struct Mol Biol. 2012;19:291–298. doi: 10.1038/nsmb.2240. Medline CrossRef. [DOI] [PubMed] [Google Scholar]
- 50.Li D, et al. pFind: a novel database-searching software system for automated peptide and protein identification via tandem mass spectrometry. Bioinformatics. 2005;21:3049–3050. doi: 10.1093/bioinformatics/bti439. Medline CrossRef. [DOI] [PubMed] [Google Scholar]
- 51.Lutz R, Bujard H. Independent and tight regulation of transcriptional units in Escherichia coli via the LacR/O, the TetR/O and AraC/I1-I2 regulatory elements. Nucleic Acids Res. 1997;25:1203–1210. doi: 10.1093/nar/25.6.1203. Medline CrossRef. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Epshtein V, Toulme F, Rahmouni AR, Borukhov S, Nudler E. Transcription through the roadblocks: the role of RNA polymerase cooperation. EMBO J. 2003;22:4719–4727. doi: 10.1093/emboj/cdg452. Medline CrossRef. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pósfai G, et al. Emergent properties of reduced-genome Escherichia coli. Science. 2006;312:1044–1046. doi: 10.1126/science.1126439. Medline CrossRef. [DOI] [PubMed] [Google Scholar]
- 54.Cardinale CJ, et al. Termination factor Rho and its cofactors NusA and NusG silence foreign DNA in E. coli. Science. 2008;320:935–938. doi: 10.1126/science.1152763. Medline CrossRef. [DOI] [PMC free article] [PubMed] [Google Scholar]