

Abstract
PGE2 is an important pro-angiogenic and pro-proliferative cytokine and the key enzymes modulating its levels, COX-2 and 15-PGDH play important opposing roles in carcinogenesis. Previously we found loss of 15-PGDH expression in lung cancer and its reactivation leads to strong in vivo tumor-suppressive effect via an antiangiogenic mechanism. Here we find that HDAC inhibitors (HDACI), such as trichostatin A (TSA) and vorinostat could reactivate 15-PGDH expression but overall induce PGE2 generation and this is the result of concomitant induction of COX-1 and 2 leading to functional promotion of endothelial cell proliferation and capillary formation. Direct TSA treatment inhibits endothelial cell proliferation and capillary formation in our study in line with prior reports as HDACIs have been shown to directly inhibit angiogenesis. The elevation of PGE2 levels induced by HDACI is potently neutralized by indomethacin (INN) or celecoxib co-treatment and accordingly, angiogenesis is more effectively inhibited when using conditioned medium of co-treatment than either alone confirming that this effect is mediated via the PGE2 axis. Accordingly, blockage of EP2/4 receptors mitigates the stimulation of angiogenesis by excessive PGE2 generation mediated by TSA. In this study, we identify a potentially adverse effect of HDACIs through induction of both 15-PGDH and COX-2 leading to elevated PGE2 levels and thereby stimulation of angiogenesis. Co-treatment of TSA and INN shows more potent anti-angiogenic effects by inducing 15-PGDH and inhibiting COX-2. Overall, our results suggest that combined HDACI and COX inhibition should be explored clinically to achieve more meaningful benefits from HDACI therapy in lung cancer.
Introduction
Lung cancer remains the leading cause of cancer deaths in the United States. Although the efficacy of systemic therapy and patient outcomes have improved in recent years, effective lung cancer treatment is hindered by the high occurrence of drug resistance, subsequent treatment failure and patient mortality, resulting in a critical need to identify and exploit novel therapeutic targets and drug combinations to improve clinical efficacy.
Prostaglandin E2 (PGE2) promotes cancer progression by affecting cell proliferation, apoptosis, angiogenesis, and the immune response via stimulating a group of four transmembrane cell surface receptors, EP1-4[1]. Cyclooxygenases (COXs), particularly inducible COX-2 convert arachidonic acid into an endoperoxide intermediate that is further metabolized to PGE2. COX-2 has been found up-regulated in many cancers and has been associated with increased VEGF production and angiogenesis. The level of PGE2 is controlled not only by synthesis but also by degradation. The key enzyme responsible for metabolic inactivation of PGE2 is NAD+-dependent 15-hydroxyprostaglandin dehydrogenase (15-PGDH) which is widely distributed in various mammalian tissues among which lung is one of the tissues with the highest level of expression[2,3]. However, 15-PGDH has been found down-regulated in various cancers including lung cancer [4-7]. In our previous study, loss of 15-PGDH expression was found in 65% of lung cancers by Western blotting of lung cancer cell lines and immunohistochemical examination of human lung cancer tissues. Further studies using in vitro cell-based assays and in vivo xenograft tumorigenesis assays showed significant in vivo tumor suppressor activity of 15-PGDH through PGE2 degradation via an antiangiogenic mechanism analogous to its role in colon cancer[8].
Angiogenesis is essential for the development and growth of tumors. HDACIs overall have been demonstrated to have a potent anti-angiogenic effect in in vitro angiogenesis assays. One class of compounds identified as HDAC inhibitors is hydroxamic acids, such as TSA and vorinostat which have demonstrated potent cytotoxicity in vitro against a variety of solid tumor cell lines[9,10]. TSA induces the expression of p53 and von Hippel-Lindau (VHL) proteins under hypoxic conditions, whereby it reduces the expression of HIF-1α and VEGF [11]. TSA and vorinostat treatment were shown to prevent vascular endothelial growth factor (VEGF)-stimulated human umbilical cord endothelial cells (HUVEC) from invading type I collagen gel and forming capillary- like structures. TSA and vorinostat also inhibit VEGF-induced formation of a CD31-positive capillary-like network in embryoid bodies and inhibit VEGF-induced angiogenesis [12]. TSA also prevents the sprouting of capillaries from rat aortic rings [12]. Vorinostat is currently FDA-approved for the treatment of cutaneous T-cell lymphoma[13] and is in clinical investigations for mesothelioma[14], non-small cell lung cancer (NSCLC) [15]and colon cancer[16]. Vorinostat enhances the efficacy of carboplatin and paclitaxel in patients with advanced NSCLC [17] and thereby HDAC inhibition is a promising therapeutic strategy for treatment of NSCLC.
In this study, we find that TSA up-regulates 15-PGDH in lung cancer cell lines. However, we also find that TSA up-regulates both COX-1 and 2 in most cell lines examined and thereby promotes angiogenesis which can be blocked by inhibition of PGE2-stimulated EP2 and EP4-receptors as well as the nonselective COX inhibitor INN and the selective COX-2 inhibitor, celecoxib suggesting that combination therapy of TSA and COX inhibitors or EP2/4 blockers can overcome this undesirable effect of HDAC inhibitor therapy and thereby presents a more powerful and beneficial treatment strategy.
Materials and Methods
Cell lines and reagents
The following normal bronchial epithelial and lung cancer cell lines, all from American Type Culture Collection, were used in this study: NHBE were grown in F-12K supplemented with 4% fetal bovine serum (FBS), H211, H23, H292, H322, H460, Calu-1 cell lines were grown in RPMI 1640 supplemented with 10% FBS. Two of these cell lines, H23 and H292 were randomly selected and confirmed as 100% genetically accurate by the STR Cell Authentication Service of ATCC. PGE2, TSA, INN and Celecoxib were purchased from Sigma Aldrich. AH6809 and L161,982 were purchased from Cayman Chemical. Vorinostat was purchased from Selleck.
Quantitative reverse transcription-PCR assay
Total RNAs were collected using RNeasy Mini kit (Qiagen) from cells with or without TSA treatment. cDNAs were synthesized with Moloney murine leukemia virus reverse transcriptase (SuperScript III reverse transcriptase) with the use of oligo(dT) primers from Invitrogen. All samples were run in triplicates on Roche LightCycler with the use of Syber green probes (Roche) using the following variables: denaturation at 95 °C for 10 min, followed by 45 cycles of amplification (95 °C 10 s, 60 °C 10 s, and 72 °C 15 s), and cooled to 40 °C at a transition rate of 20 °C/s. Levels of glyceraldehyde-3-phosphate dehydrogenase expression were used as internal reference to normalize input cDNA. Ratios of level of each gene to glyceraldehyde-3-phosphate dehydrogenase were then calculated. Primer sequences are available upon request.
Immunoblotting
Whole cell lysates were isolated using radio immunoprecipitation assay buffer (Upstate Biotechnology) supplemented with protease inhibitor mixture (Roche Applied Sciences) and 50 μg of protein/lane were electrophoresed in 8% SDS-PAGE minigels. A dilution of 1:1,000 monoclonal mouse anti-15-PGDH antibody (provided by Dr. Sanford D. Markowitz, Case Western Reserve University), 1:1,000 polyclonal goat anti-Cox-2 antibody (Santa Cruz Biotechnology), 1:5000 total ERK and phosphorylated ERK (Cell signaling) and 1:5,000 polyclonal rabbit GAPDH antibody (Cell Signaling) were used. Detection was performed using Western Lightning Chemiluminescence reagent (Perkin-Elmer Life Sciences).
PGE2 ELISA
Six lung cancer cell lines were seeded in 6-well plates at a density of 3×105 cells/well and treated with or without TSA, vorinostat, INN and Celecoxib. Culture medium was collected 24 h later and centrifuged briefly to remove cell debris. PGE2 levels were examined by ELISA according to the manufacturer's instructions (Thermo Scientific). The absorbance at 405 and 590 nm was read immediately on a microplate autoreader (EL311; BIO-TEK Instruments, Inc.). Each reading of absorbance at 405 nm was normalized by the reading of absorbance at 590 nm and the concentrations of PGE2 from each sample were calculated according to the standard curve generated from the PGE2 standards provided.
Endothelial cell proliferation assay
A total of 4×103 cells in 100 μl of basal medium with 1% FBS were seeded into each well of a 96-well plate, pre-treated for 1 hour with or without 1μmol AH6809 and/or 1μmol L161,982 or vehicle control and then treated with 10% of the respective conditioned medium as the stimulant, and incubated at 37 °C for 72 h; control cells were incubated in basal medium and 1% FBS, as previously described [18]. No additional proliferation stimulus (VEGF or basic fibroblast growth factor) was added. MTS cell growth assays were performed 72 h after the addition of conditioned medium. CellTiter 96Aqueous One Solution reagent (20 μl) was added to each well and after 2-hour incubation at 37 °C, absorbance at 490 nm was determined for each well with a microplate reader (Dynex Technologies). The data presented are the average of quadruplicate experiments.
Matrigel tube formation assay
The matrigel tube formation assay was performed as previously described [19]. Briefly, each well of a prechilled 48-well cell culture plate was coated with 100 μL of unpolymerized Matrigel (BD Matrigel Matrix) and incubated at 37 °C for 30 to 45 min Human umbilical vascular endothelial cells (HUVECs) were harvested with trypsin, and 4×104 cells were resuspended in 300μl of endothelial cell basal medium supplemented with 1% FBS, and treated with 10% of the appropriate conditioned medium before plating onto the Matrigel-coated plates. After 12 h of incubation at 37 °C, endothelial cell tube formation was assessed with an inverted photomicroscope (Nikon). Microphotographs of the center of each well at low power were taken at 40× magnification with the aid of imaging-capture software (NIS-Elements from Nikon). Tube formation in the microphotographs was quantitatively analyzed (total tube length); controls consisted of human umbilical vascular endothelial cells in basal medium supplemented with 1% FBS. These experiments were done in triplicates and the data presented represent mean results.
Statistical analysis
Statistical significance was checked by two-sample t-tests, with P-value of less than 0.05 considered statistically significant. Similar conclusions were reached using non-parametric Mann Whitney U-test. Data are expressed as mean+±SD. The data are representative of three separate experiments.
Results
Induction of 15-PGDH expression in lung cancer cells treated with TSA
Down-regulation of the tumor suppressor, 15-PGDH has been found in gastric, colorectal, breast and prostate cancers and is at least in part mediated by epigenetic silencing. Down-regulation of 15-PGDH is also found in lung cancer but the mechanism of silencing remains unclear. We hypothesized that it could be mediated by epigenetic mechanisms, such as histone acetylation. To test this, we treated lung cancer cell lines with the HDACI TSA for 24 hours. Six lung cancer cell lines with no 15-PGDH expression were selected: H211, H23, H292, H322, Calu-1 and H460. A dramatic increase in 15-PGDH mRNA was found in all these six cell lines treated with 0.3uM TSA. Interestingly, TSA not only induced 15-PGDH in lung cancer cells but also in normal bronchial/tracheal epithelial cells (NHBE) (Figure1a). To further validate the induction of 15-PGDH identified by quantitative RT-PCR on the protein level, we analyzed the change in expression of 15-PGDH at 24 hours post-TSA treatment when compared to vehicle-treated control in Western blotting assays. The pattern of expression obtained for 15-PGDH showed consistent up-regulation in five out of six lung cancer cell lines tested with the exception of H322 where no protein expression was noted at all, possibly related to expression being below the level of detection with the antibody used(Figure 1b).
Figure 1.


Expression of 15-PGDH is induced by TSA in lung cancer and normal bronchial epithelial cells. A. Quantitative RT-PCR of 15-PGDH expression in NHBE, H211, H23, H292, H322, Calu1 and H460 cells following 24 hours of TSA treatment. B. Immunoblotting of 15-PGDH in lung cancer cells following 24 hours of TSA treatment. (* p<0.05; ** p<0.01; *** p<0.001 ).
Prostaglandin E2 production increased in lung cancer cells treated with TSA
15-PGDH has been considered the key enzyme responsible for the biological inactivation of prostaglandins and therefore one might expect that PGE2 would be reduced by 15-PGDH expression induced by TSA treatment. To examine the association between the induction of 15-PGDH by TSA and prostaglandin degradation, we measured the levels of PGE2 in the medium from H211 and H23 cells cultured for 12h and 24h following TSA treatment. Contradictory to our expectation, PGE2 was induced significantly in a time dependent manner (Figure 2a). We further measured the levels of PGE2 in the medium from multiple other lung cancer cell lines- H292, H322, H460 and Calu1 upon TSA treatment. Overall, 4/6 cell lines examined similarly showed PGE2 induction while in H322 a significant reduction of PGE2 was seen (Figure 2b).
Figure 2.


TSA induces PGE2 in lung cancer cell lines. A. The induction of PGE2 is time-dependent and increases as least until 24 hours of TSA treatment in H211 and H23 cells. B. PGE2 is also induced by TSA in H292 and H460 but not in Calu3 and H322 cells. (* p<0.05; ** p<0.01; *** p<0.001 ).
Increased expression of COX-1/2 in lung cancer cells and bronchial epithelial cells treated with TSA and Vorinostat
To investigate the mechanism of induction of PGE2 after HDACI treatment we first examined the effect of TSA on the induction of COX-1/2 expression in H211 and H23 cells by quantitative RT-PCR. The results showed that TSA markedly induced the expression of COX-1/2 in these two lung cancer cell lines and also in NHBE cells at the transcriptional level (Figure 3a). To further validate the induction identified by quantitative RT-RCR, we analyzed the change in expression of COX-2 at 6, 12 and 24 hours post-TSA treatment when compared to vehicle-treated control by Western blotting in H211 and H23 cell lines. We found that COX-2 expression was up-regulated in a time-dependent (Figure 3b I) and dose-dependent manner (II) and could be induced by TSA at as low as 50 nM concentration and as early as after 6 hours of treatment. Similar results were obtained with the alternative HDACI, vorinostat which shows the induction of COX2 in a dose dependent manner (III). We confirmed the induction of COX-1/2 on the protein level by TSA in all lung cancer cell lines examined (IV). These findings were also reproducible and indeed even more pronounced under hypoxic conditions that might mimic in vivo conditions better (Figure 3C I). The hypoxia-induced transcription factor, HIF1α was analysed by quantitative RT-PCR and as expected was induced during hypoxic conditions (Figure 3c II).
Figure 3.



Expression of COX-1/2 is induced by TSA and vorinostat in lung cancer and bronchial epithelial cells. A. Quantitative RT-PCR of COX-1/2 expression in H211, H23 and NHBE cells after 24 hours of TSA treatment. B. Immunoblotting of COX-2 in H211 and H23 shows that COX2 induction by TSA is time dependent (I) and dose dependent (II). Vorinostat treatment leads to analogous Cox-2 induction (III). COX1/2 induction by TSA is also observed in H292, H322, Calu-1 and H460 cells (IV) C. COX-2 induction by TSA is also observed under both normoxic and hypoxic culture conditions (I). Quantitative real-time PCR confirm that HIF-1α is induced in H23 cells during hypoxic culture condition (II). (* p<0.05; ** p<0.01; *** p<0.001 ).
INN and Celecoxib neutralize the induction of PGE2 by TSA and vorinostat
In order to further corroborate that PGE2 induction is dependent on COX-1/2 induction, we cultured H211 and H23 cells for 24 h in the presence or absence of TSA (300nM), vorinostat (500nM) and/or the COX inhibitor, indomethacin (INN, 10μM) co-treatment and the amount of PGE2 released into the medium was determined. As shown in Figure 4a, at the non-toxic dose of 10μM, INN potently neutralized the induction of PGE2 by both TSA and vorinostat to levels lower than control cells. We treated H211 and H23 with or without TSA and/or the selective COX-2 inhibitor celecoxib and find that 1μM of the COX-2 selective inhibitor, celecoxib could neutralize the induction of PGE2 as potently as INN suggesting that COX-2 plays a more important role in the up regulation of PGE2 than COX-1 (Figure 4b).
Figure 4.


Combined treatment of HDACIs for 24h along with COX inhibitors neutralizes the induction of PGE2 by TSA in H211 and H23 cells. A. Analysis of PGE2 level by ELISA after combined treatment with TSA/vorinostat and INN for 24 hours in H211 and H23 cells B. Analysis of PGE2 by ELISA after combined treatment with TSA and celecoxib for 24 hours in H211 and H23 cells. (* p<0.05; ** p<0.01; *** p<0.001 ).
Anti-angiogenic effect of combined therapy of TSA and INN
The induction of PGE2 by TSA noted in human lung cancer cells suggests that TSA might paradoxically contribute to the promotion of angiogenesis in lung tumors in addition to its known direct anti-angiogenic effect. To test this hypothesis, we conducted MTS cell growth assays with HUVEC cells with the addition of conditioned medium from H23 cells treated with or without TSA and other inhibitors. First we confirmed the expression of the PGE2-stimulated EP2 and EP4 receptor by Western blot in HUVEC cells. Then, we starved HUVEC cells with basal medium overnight and then pretreated cells for one hour with or without EP2 antagonist (1μM AH6809) or EP4 receptor antagonist (1μM L-161,982), followed by treatment for 72 hours with conditioned medium. The conditioned medium was obtained by collecting the culture medium of H23 cells treated with TSA and diluted into 10% by basal medium while the control group received conditioned medium of H23 cells that were not treated with TSA (Figure 5a) showed that the conditioned media of H23 cells treated with TSA could stimulate HUVEC cell proliferation and that both EP2 and EP4 antagonist could potently block this effect of TSA. To demonstrate appropriate receptor blockade by these inhibitors, we performed ERK signaling studies with HUVEC cells and show that AH6809 and L161,982 potently block Erk signaling activation by PGE2 (Figure 5b). As shown in Figure 4, INN could potently neutralize the induction of PGE2 by TSA. Therefore, we starved HUVEC cells and then added different conditioned medium obtained by collecting the cultured medium of H23 cells treated with or without TSA and/or INN. As shown in Figure 5c, the conditioned medium of H23 cells treated with TSA and INN not only blocked the stimulation of HUVEC cell proliferation to the level expected as a result of TSA treatment but also the presence of INN further inhibited HUVEC cell growth suggesting the important role PGE2 might play in endothelial cell proliferation. In order to further prove the pro-angiogenic effect of TSA on lung cancer cells, we conducted matrigel tube formation assays with the respective conditioned medium used in the MTS cell growth assay. Figure 6 shows the reduced capillary tube formation of HUVEC cells directly treated with TSA and the induced capillary tube formation of HUVEC cells grown in the presence of conditioned medium of H23 treated with TSA. Treatment with the conditioned medium of H23 cells treated with the combination of TSA and INN could inhibit angiogenesis even more effectively than either alone suggestive of the clinical benefit of such a combination strategy to optimize the use of HDAC inhibitor therapy for the treatment of lung cancer.
Figure 5.


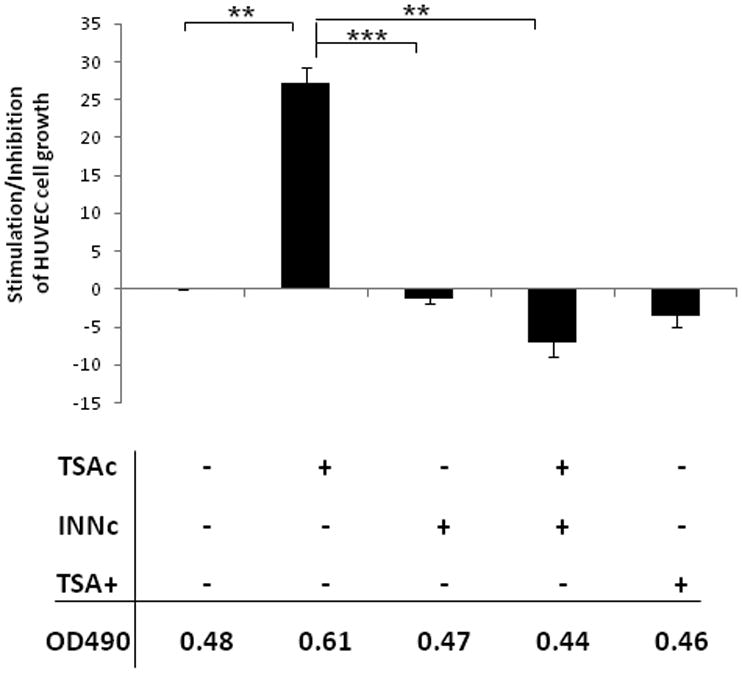
The stimulation of HUVEC cell growth by TSA is PGE2 mediated. A. MTS assay of HUVEC cell growth for 72 hours after pretreatment with EP2 (AH6809) or EP4 (L161,982) antagonists for 1 hour, followed by the addition of 10% conditioned medium- i.e. collected culture medium of H23 cells treated with TSA for 24 hours. B. Western blot shows that EP2 and EP4 receptors are strongly expressed in HUVEC cells. Both AH6809 and L161,982 treatment potently block ERK activation after PGE2 stimulation. C. MTS assay of HUVEC cell growth following treatment with different conditioned media. TSAc is the collected medium of H23 cells treated with TSA for 24 hours. INNc is the collected medium of H23 cells treated with INN for 24 hours. TSA+ is the direct treatment of TSA of HUVEC cells. (* p<0.05; ** p<0.01; *** p<0.001 ).
Figure 6.

Effect of PGE2 induced by TSA on tumor angiogenesis. Matrigel tube formation assays show that direct TSA treatment by itself (TSA+) could reduce capillary tube formation of HUVEC cells while conversely the conditioned medium of TSA treated H23 cells (TSAc) could promote angiogenesis. The conditioned medium of H23 cells treated with the combination of INN and TSA (TSA+INNc) shows more significant reduction of capillary tube formation of HUVEC cells compared with other groups. Quantification is achieved by measurement of the length of vessel-like extensions from the explants. (* p<0.05; ** p<0.01; *** p<0.001 ).
Discussion
The oncogene, COX-2 activity generates PGE2 inducing tumor-associated angiogenesis and PGE2 is metabolized by the 15-PGDH enzyme. Previously, we showed the tumor suppressor activity of 15-PGDH via an anti-angiogenic mechanism in lung cancer. One of the key objectives of this study was to identify COXs and 15-PGDH which were consistently regulated by TSA in lung cancer cell lines examined. Down-regulation of 15-PGDH has been found in lung cancers and might be in part mediated by epigenetic silencing. Epigenetic strategies, in particular HDACs have shown promise in combination with chemotherapy in the treatment of advanced non-small cell lung cancer. We hypothesized that the down regulation of 15-PGDH in lung cancer is also mediated by epigenetic silencing and therefore we treated lung cancer cell lines with HDAC inhibitors, such as TSA and vorinostat in order to reverse epigenetic silencing. While 15-PGDH re-activation can be detected with TSA treatment in lung cancer cell lines, however, TSA was found to markedly enhance PGE2 generation via the induction of COX-1/2 expression in the majority lung cancer cell lines we studied. In addition, most lung cancer cell lines produced significantly more PGE2 than untreated cells following TSA treatment, indicating that the induced COX enzymes are functionally active. To confirm that PGE2 production was associated with the catalytic activity of COXs, PGE2 levels were measured after co-treatment with TSA and INN. Interestingly, non-cytotoxic, 10μM concentration of INN potently blocked PGE2 production induced by TSA, namely decreased PGE2 to the level of the control group in the cell lines examined. The concentrations of INN which caused cell death in lung cancer cell lines (>200μM) [20] far exceeded the concentrations required to inhibit cyclooxygenase activity (<10μM) implying the involvement of apoptosis independent mechanisms in the inhibition of HUVEC cell growth. Interestingly, TSA up-regulated COX1/2 in normal bronchial/tracheal epithelial cells as well suggesting that utility of combining HDACI and COX inhibitor therapy might be more broadly meaningful as similar adverse events by HDACI therapy might be elicited from normal tissues as well. This is even more relevant as the inflammatory mediator PGE2 is increased in the lungs of COPD patients [21] and enhanced levels of PGE2 correlate with the severity of airflow limitation in stable chronic obstructive pulmonary disease (COPD) [22]. Loss of HDACs, in particular HDAC2 is noted in patients with worsening COPD and this loss is associated with the induction of pro-inflammatory proteins/cytokines, such as IL-8, COX-2, PGE2 [23]. This suggests that HDAC inhibitors could potentially have detrimental effects on normal lung tissue and such detrimental effects could also be ameliorated by the concurrent use of anti-inflammatory drugs, such as COX inhibitors. COX-1 and COX-2 are key enzymes in the biosynthetic pathway by which arachidonic acid is converted into an intermediate prostaglandin, PGG2 and then metabolized to PGE2. COX-1 is expressed at relatively constant levels in numerous tissues, as compared to the expression of COX-2 which is normally low or absent in most cells and tissues but rapidly up-regulated by tumor promoters[24]. Thus, COX-2 represents the better target for therapeutic intervention in cancer. Celecoxib inhibits COX-2 without affecting COX-1[25], therefore we measured PGE2 levels after co-treatment with TSA and Celecoxib. Celecoxib neutralized the up-regulation of PGE2 by TSA as effectively as INN suggesting that COX-2 serves as a more important mediator in the pathway. TSA could induce 15-PGDH and both COX enzymes on both the transcriptional and translational level in lung cancer cell lines. The induction of COX1/2 was more dramatic under tumor hypoxic conditions and HIF-1α was induced as expected. However, the up-regulation of PGE2 via the induction of COX-2 outweighs its down-regulation via the induction of 15-PGDH. The up-regulation of PGE2 by TSA could promote endothelial cell proliferation and capillary formation. In order to investigate whether the promotion of angiogenesis by PGE2 is VEGF-dependent, we measured the levels of VEGF after TSA treatment and found that TSA could down-regulate VEGF levels as expected (data not shown) suggesting the existence of another important pro-angiogenic factor which outweighs the effect on VEGF. It has previously been reported that PGE2 itself stimulates angiogenesis [26]. PGE2 induces angiogenesis via activation of EP2 and EP4 receptors in human vascular endothelial cells. EP2 or EP4 activation induces an increase of intracellular levels of cAMP and ERK1/2, linked to different signaling pathways. EP2 and EP4 antagonist potently blocked both the pro-angiogenic effect on HUVEC cells of conditioned medium obtained from TSA-treated lung cancer cells as well as PGE2-stimulated Erk1/2 signaling suggesting that the stimulation of proliferation is indeed PGE2 mediated and dependent on the stimulation of EP2/4-receptor driven pathways.
HDACI are a new class of promising anti-tumor agents inhibiting angiogenesis with very low toxicity toward normal cells [27, 28]. TSA, a HDACI alters gene expression by interfering with the removal of acetyl groups from histones and therefore altering the ability of DNA-binding transcription factors to access the DNA molecules inside chromatin thereby affecting the transcription of genes, including tumor suppressor genes and oncogenes. Several studies to date have demonstrated that HDACI induce alterations in the expression of multiple drug targets and/or metabolic pathways that are critical molecular determinants for cancer therapeutics. Importantly, combination treatment with additional agents targeting these modulated pathways has resulted in synergistic growth inhibitory effects on cancer cells in vitro and in vivo. HDACI have shown preclinical efficacy in solid tumors, including ovarian cancers. A recent study reported that the combined treatment of the HDACI and COXs inhibitor showed enhanced anti tumor effects in ovarian cancer cells [29].
In this study, we demonstrate that TSA treatment has paradoxical effects in lung cancer: besides its direct anti-angiogenic effects, it up-regulates 15-PGDH but still exerts a dominant pro-angiogenic effect via the concomitant prominent up-regulation of COX1/2. We show that the anti-angiogenic function of TSA can be enhanced by blocking its pro-angiogenic function via co-treatment with the COX inhibitors, INN and celecoxib. Our study identified the PGE2 pathway as an important determinant for angiogenesis in lung cancer and suggests COX-2 and 15-PGDH expression as potential candidate biomarkers for HDACI-based treatment for lung cancer. In conclusion, 15-PGDH and COX-2 are inducible genes in lung cancer cells and appear to be transcriptionally regulated by a mechanism associated with histone acetylation. HDACIs are a new class of promising anti-tumor agents inhibiting cell proliferation and survival in tumor cells with very low toxicity toward normal cells. Induction of COXs and as a result increased PGE2 production by HDAC inhibition is an untoward side effect of clinical relevance. INN/Celecoxib may not only functionally neutralize the effect produced by TSA but also shows more potent anti-angiogenic effects when used together with TSA revealing potent anti angiogenic properties of the combination of HDACI and COX inhibition in lung cancer.
Footnotes
Xiaoqi Wang and Guangyuan Li contributed equally.
References
- 1.Holla VR, Backlund MG, Yang P, Newman RA, DuBois RN. Regulation of prostaglandin transporters in colorectal neoplasia. Cancer Prev Res (Phila Pa) 2008;1(2):93–99. doi: 10.1158/1940-6207.CAPR-07-0009. [DOI] [PubMed] [Google Scholar]
- 2.Tai HH, Cho H, Tong M, Ding Y. NAD+-linked 15-hydroxyprostaglandin dehydrogenase: structure and biological functions. Curr Pharm Des. 2006;12(8):955–962. doi: 10.2174/138161206776055958. [DOI] [PubMed] [Google Scholar]
- 3.Anggard E, Larsson C, Samuelsson B. The distribution of 15-hydroxy prostaglandin dehydrogenase and prostaglandin-delta 13-reductase in tissues of the swine. Acta Physiol Scand. 1971;81(3):396–404. doi: 10.1111/j.1748-1716.1971.tb04914.x. [DOI] [PubMed] [Google Scholar]
- 4.Moreno J, Krishnan AV, Swami S, Nonn L, Peehl DM, Feldman D. Regulation of prostaglandin metabolism by calcitriol attenuates growth stimulation in prostate cancer cells. Cancer Res. 2005;65(17):7917–7925. doi: 10.1158/0008-5472.CAN-05-1435. [DOI] [PubMed] [Google Scholar]
- 5.Myung SJ, Rerko RM, Yan M, et al. 15-Hydroxyprostaglandin dehydrogenase is an in vivo suppressor of colon tumorigenesis. Proc Natl Acad Sci U S A. 2006;103(32):12098–12102. doi: 10.1073/pnas.0603235103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wolf I, O'Kelly J, Rubinek T, et al. 15-hydroxyprostaglandin dehydrogenase is a tumor suppressor of human breast cancer. Cancer Res. 2006;66(15):7818–7823. doi: 10.1158/0008-5472.CAN-05-4368. [DOI] [PubMed] [Google Scholar]
- 7.Backlund MG, Mann JR, Holla VR, et al. 15-Hydroxyprostaglandin dehydrogenase is down-regulated in colorectal cancer. J Biol Chem. 2005;280(5):3217–3223. doi: 10.1074/jbc.M411221200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang G, Eisenberg R, Yan M, et al. 15-Hydroxyprostaglandin dehydrogenase is a target of hepatocyte nuclear factor 3beta and a tumor suppressor in lung cancer. Cancer Res. 2008;68(13):5040–5048. doi: 10.1158/0008-5472.CAN-07-6575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Richon VM, Emiliani S, Verdin E, et al. A class of hybrid polar inducers of transformed cell differentiation inhibits histone deacetylases. Proc Natl Acad Sci U S A. 1998;95(6):3003–3007. doi: 10.1073/pnas.95.6.3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Richon VM, Garcia-Vargas J, Hardwick JS. Development of vorinostat: current applications and future perspectives for cancer therapy. Cancer Lett. 2009;280(2):201–210. doi: 10.1016/j.canlet.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 11.Kim MS, Kwon HJ, Lee YM, et al. Histone deacetylases induce angiogenesis by negative regulation of tumor suppressor genes. Nat Med. 2001;7(4):437–443. doi: 10.1038/86507. [DOI] [PubMed] [Google Scholar]
- 12.Deroanne CF, Bonjean K, Servotte S, et al. Histone deacetylases inhibitors as anti-angiogenic agents altering vascular endothelial growth factor signaling. Oncogene. 2002;21(3):427–436. doi: 10.1038/sj.onc.1205108. [DOI] [PubMed] [Google Scholar]
- 13.Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist. 2007;12(10):1247–1252. doi: 10.1634/theoncologist.12-10-1247. [DOI] [PubMed] [Google Scholar]
- 14.Paik PK, Krug LM. Histone deacetylase inhibitors in malignant pleural mesothelioma: preclinical rationale and clinical trials. J Thorac Oncol. 5(2):275–279. doi: 10.1097/JTO.0b013e3181c5e366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Traynor AM, Dubey S, Eickhoff JC, et al. Vorinostat (NSC# 701852) in patients with relapsed non-small cell lung cancer: a Wisconsin Oncology Network phase II study. J Thorac Oncol. 2009;4(4):522–526. doi: 10.1097/jto.0b013e3181952478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pitts TM, Morrow M, Kaufman SA, Tentler JJ, Eckhardt SG. Vorinostat and bortezomib exert synergistic antiproliferative and proapoptotic effects in colon cancer cell models. Mol Cancer Ther. 2009;8(2):342–349. doi: 10.1158/1535-7163.MCT-08-0534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ramalingam SS, Maitland ML, Frankel P, et al. Carboplatin and Paclitaxel in combination with either vorinostat or placebo for first-line therapy of advanced non-small-cell lung cancer. J Clin Oncol. 28(1):56–62. doi: 10.1200/JCO.2009.24.9094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Merchan JR, Chan B, Kale S, Schnipper LE, Sukhatme VP. In vitro and in vivo induction of antiangiogenic activity by plasminogen activators and captopril. J Natl Cancer Inst. 2003;95(5):388–399. doi: 10.1093/jnci/95.5.388. [DOI] [PubMed] [Google Scholar]
- 19.Merchan JR, Jayaram DR, Supko JG, He X, Bubley GJ, Sukhatme VP. Increased endothelial uptake of paclitaxel as a potential mechanism for its antiangiogenic effects: potentiation by Cox-2 inhibition. Int J Cancer. 2005;113(3):490–498. doi: 10.1002/ijc.20595. [DOI] [PubMed] [Google Scholar]
- 20.Sanchez-Alcazar JA, Bradbury DA, Pang L, Knox AJ. Cyclooxygenase (COX) inhibitors induce apoptosis in non-small cell lung cancer through cyclooxygenase independent pathways. Lung Cancer. 2003;40(1):33–44. doi: 10.1016/s0169-5002(02)00530-5. [DOI] [PubMed] [Google Scholar]
- 21.Profita M, Sala A, Bonanno A, et al. Chronic obstructive pulmonary disease and neutrophil infiltration: role of cigarette smoke and cyclooxygenase products. Am J Physiol Lung Cell Mol Physiol. 298(2):L261–269. doi: 10.1152/ajplung.90593.2008. [DOI] [PubMed] [Google Scholar]
- 22.Chen Y, Chen P, Hanaoka M, Droma Y, Kubo K. Enhanced levels of prostaglandin E2 and matrix metalloproteinase-2 correlate with the severity of airflow limitation in stable COPD. Respirology. 2008;13(7):1014–1021. doi: 10.1111/j.1440-1843.2008.01365.x. [DOI] [PubMed] [Google Scholar]
- 23.Ito K, Ito M, Elliott WM, et al. Decreased histone deacetylase activity in chronic obstructive pulmonary disease. N Engl J Med. 2005;352(19):1967–1976. doi: 10.1056/NEJMoa041892. [DOI] [PubMed] [Google Scholar]
- 24.Herschman HR. Prostaglandin synthase 2. Biochim Biophys Acta. 1996;1299(1):125–140. doi: 10.1016/0005-2760(95)00194-8. [DOI] [PubMed] [Google Scholar]
- 25.Fort J. Celecoxib, a COX-2--specific inhibitor: the clinical data. Am J Orthop (Belle Mead NJ) 1999;28(3 Suppl):13–18. [PubMed] [Google Scholar]
- 26.Alfranca A, Lopez-Oliva JM, Genis L, et al. PGE2 induces angiogenesis via MT1-MMP-mediated activation of the TGFbeta/Alk5 signaling pathway. Blood. 2008;112(4):1120–1128. doi: 10.1182/blood-2007-09-112268. [DOI] [PubMed] [Google Scholar]
- 27.Muhlethaler-Mottet A, Meier R, Flahaut M, et al. Complex molecular mechanisms cooperate to mediate histone deacetylase inhibitors anti-tumour activity in neuroblastoma cells. Mol Cancer. 2008;7:55. doi: 10.1186/1476-4598-7-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ellis L, Pili R. Histone Deacetylase Inhibitors: Advancing Therapeutic Strategies in Hematological and Solid Malignancies. Pharmaceuticals(Basel) 3(8):2411–2469. doi: 10.3390/ph3082441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Son DS, Wilson AJ, Parl AK, Khabele D. The effects of the histone deacetylase inhibitor romidepsin (FK228) are enhanced by aspirin (ASA) in COX-1 positive ovarian cancer cells through augmentation of p21. Cancer Biol Ther. 9(11):928–935. doi: 10.4161/cbt.9.11.11873. [DOI] [PMC free article] [PubMed] [Google Scholar]