

Abstract
Apaf-1−/− or caspase-3−/− cells treated with a variety of apoptosis inducers manifest apoptosis-associated alterations including the translocation of apoptosis-inducing factor (AIF) from mitochondria to nuclei, large scale DNA fragmentation, and initial chromatin condensation (stage I). However, when compared with normal control cells, Apaf-1−/− or caspase-3−/− cells fail to exhibit oligonucleosomal chromatin digestion and a more advanced pattern of chromatin condensation (stage II). Microinjection of such cells with recombinant AIF only causes peripheral chromatin condensation (stage I), whereas microinjection with activated caspase-3 or its downstream target caspase-activated DNAse (CAD) causes a more pronounced type of chromatin condensation (stage II). Similarly, when added to purified HeLa nuclei, AIF causes stage I chromatin condensation and large-scale DNA fragmentation, whereas CAD induces stage II chromatin condensation and oligonucleosomal DNA degradation. Furthermore, in a cell-free system, concomitant neutralization of AIF and CAD is required to suppress the nuclear DNA loss caused by cytoplasmic extracts from apoptotic wild-type cells. In contrast, AIF depletion alone suffices to suppress the nuclear DNA loss contained in extracts from apoptotic Apaf-1−/− or caspase-3−/− cells. As a result, at least two redundant parallel pathways may lead to chromatin processing during apoptosis. One of these pathways involves Apaf-1 and caspases, as well as CAD, and leads to oligonucleosomal DNA fragmentation and advanced chromatin condensation. The other pathway, which is caspase-independent, involves AIF and leads to large-scale DNA fragmentation and peripheral chromatin condensation.
Keywords: apoptosis-inducing factor, Apaf-1, chromatin condensation, caspases, caspase-activated DNase
Introduction
One of the hallmarks of apoptosis is the degradation and concomitant compaction of chromatin. It has been generally assumed that caspases as well as downstream effectors such as caspase-activated DNase (CAD) and Acinus are rate limiting for the development of nuclear apoptosis 1 2 3 4 5. Accordingly, the inactivation of the caspase-3 gene 6 7 and that of the caspase activator Apaf-1 8 can delay cell death and largely abolish the type of chromatin condensation observed in normal control cells treated with apoptosis inducers such as staurosporine (STS) or etoposide. Similarly, the inactivation of CAD prevents advanced chromatin condensation in different cell types 9. However, caspases and CAD are not the only effectors causing nuclear apoptosis. Thus, chromatin condensation has been observed in lymphoid cells treated with STS, anti-CD2 10, anti-CD4, or anti-CXCR4 11, as well as in fibroblasts overexpressing PML 12, even when caspase activation is inhibited. Partial chromatin condensation is found in thymocytes undergoing apoptosis in the presence of the pan-caspase inhibitor Z-VAD.fmk 13. Recently, apoptosis-inducing factor (AIF), a mitochondrial intermembrane flavoprotein, has been found to translocate from mitochondria to nuclei in a caspase-independent fashion. When added to purified nuclei, recombinant AIF causes caspase-independent large scale (∼50 kb) DNA fragmentation and a type of peripheral chromatin condensation that resembles the first stage of nuclear apoptosis (stage I) observed in intact cells undergoing apoptosis 14 15. Here, we examined the question of whether several independent pathways may lead to nuclear apoptosis.
Materials and Methods
Cells and Microinjection.
Mouse embryonic fibroblasts (MEFs) obtained from caspase 3−/− 7, Apaf-1−/− 8, or control mice were cultured with STS (2 μM), etoposide (100 μM), cisplatin (150 μM), arsenite (50 μM; Sigma-Aldrich), and/or Z-VAD.fmk (50 μM; Enzyme Systems). Cells were microinjected (pressure 150 hPa; 0.4 s; reference 16) with the following: buffer only; recombinant AIF; an inactive deletion mutant of AIF (Δ1-351; reference 14); horse cytochrome c (Cyt-c; Sigma-Aldrich); recombinant active caspase-3 17; or inactive inhibitor of CAD (ICAD)/CAD or active CAD (generated by digestion of the 250 nM ICAD–CAD complex with 3 U of caspase-3 in 10 μM of CAD buffer; 30 min at room temperature, followed by addition of 100 μM Ac-DEVD.fmk; Enzyme systems). After microinjection, cells were cultured for 180 min and stained for 10 min with the mitochondrial transmembrane potential (ΔΨm)-sensitive dye CMXRos (100 nM) and the DNA-intercalating dye Hoechst 33342 (1.5 μM; reference 16). Microinjected viable cells (100 per session; two to three independent sessions of injection) were identified by inclusion of 0.25% (wt/vol) FITC-dextran (green fluorescence) in the injectate. Only the blue and red fluorescence was recorded.
Immunofluorescence Staining.
Fixed and permeabilized MEFs were stained for AIF and Cyt-c as described 14 15. A rabbit polyclonal antiserum, CM1 (revealed as anti-AIF), was used to detect the p18 subunit of cleaved caspase-3 18. Unfixed cells were incubated for 15 min with 1.2 μM ΔΨm-sensitive JC-1 (Molecular Probes). Confocal microscopy was performed on a Leica TC-SP equipped with an ArKr laser mounted on an inverted Leica DM IFBE microscope with an 63 × 1.32 numerical aperture oil objective. Two stages of nuclear apoptosis were distinguished by staining with 25 nM Sytox-green (Molecular Probes) for 15 min at room temperature. Stage I was characterized by rippled nuclear contours and a rather partial chromatin condensation, and stage II by a more pronounced pattern of chromatin condensation 14 15.
Electron Microscopy.
Cells were fixed in pellets for 1 h at 4°C with 1.6% glutaraldehyde in 0.1 M sodium phosphate buffer, pH 7.4, washed three times, and then post-fixed in 1% osmic acid in phosphate buffer before scrapping, dehydration, and embedding. Ultrathin sections mounted on 200 mesh grids were examined in a JEOL 1200 EX electron microscope.
DNA Gel Electrophoresis.
Oligonucleosomal DNA fragmentation was detected by agarose gel electrophoresis 19. For pulse field gel electrophoresis, DNA was prepared from agarose plugs (106 cells; reference 20) and analyzed in a Bio-Rad CHEF-DR II (1% agarose; TBE; 200 V; 24 h; pulse wave 60 s; 120° angle; Bio-Rad Laboratories).
Cell-free Systems of Nuclear Apoptosis.
Cytosols from MEFs stimulated for 24 h with STS (2 μM), etoposide (100 μM), or cisplatin (150 μM) were prepared in cell-free system buffer (50 μl/106 cells) supplemented with 50 μM Z-VAD.fmk, as previously described 21. Immunodepletion of AIF (or sham immunodepletion) was performed using an anti-AIF antiserum (or preimmune serum) and protein A/G coupled to agarose (Santa-Cruz Biotechnology, Inc.; reference 14). Purified HeLa cell nuclei were exposed to cytolic extracts (2 μg/μl protein), AIF 14, CAD, ICAD 4, caspase-3 17, and/or the AIF inhibitor para-chloromercuriphenylsulfonic acid (PCMPS; Sigma-Aldrich; reference 22) in cell-free system buffer 23, and nuclear DNA content was quantitated by cytofluorometry after staining with propidium iodide 14.
Results and Discussion
Mitochondrial Membrane Permeabilization, Initial Nuclear Apoptosis, and Large Scale DNA Fragmentation Occur in Apaf-1−/− and caspase-3−/− Cells.
Apaf-1−/− or caspase-3−/− cells, as well as control MEF, responded to four different apoptosis inducers (STS, etoposide, cisplatin or arsenite) by manifesting a decrease in the ΔΨm, translocation of AIF from mitochondria to nuclei, and translocation of Cyt-c from mitochondria to the cytoplasm (Fig. 1A and Fig. B). As expected, at no time point did Apaf-1−/− or caspase-3−/− MEFs stain with an antibody specific for activated caspase-3 (Fig. 1 A). The nuclear phenotype manifested by Apaf-1−/− or caspase-3−/− cells appeared clearly distinct from that of control cells. Apaf-1−/− or caspase-3−/− cells (Fig. 1A Fig. c) only manifested a minor peripheral chromatin condensation (stage I), as was also found in control cells after short-term incubation with STS. However, Apaf-1−/− or caspase-3−/− cells failed to develop the more advanced chromatin condensation (stage II) of control cells (Fig. 1A Fig. c). None of the mitochondrial parameters nor the pattern or kinetics of chromatin condensation of Apaf-1−/− or caspase-3−/− cells (Fig. 1A and Fig. B) were influenced by addition of the pan-caspase inhibitor Z-VAD.fmk. However, Z-VAD.fmk arrested the nuclear apoptosis of control wild-type cells at stage I (Fig. 1A and Fig. B). At the ultrastructural level, nuclei from apoptotic Apaf-1−/− or caspase-3−/− cells demonstrated a rather partial chromatin condensation, with patches of chromatin abutting to the apparently intact envelope and without nucleolar degradation (Fig. 1 C). As a biochemical correlation of the morphological features of caspase-independent apoptosis, Apaf-1−/− or caspase-3−/− cells developed large scale DNA fragmentation to ∼50 kb (Fig. 1 D), yet failed to show the (presumably CAD-mediated; references 1, 2, 9) oligonucleosomal DNA fragmentation (Fig. 1 E).
Figure 1.
Mitochondrial and nuclear features of apoptosis in Apaf-1−/− and caspase-3−/− cells. MEFs from control mice (wild-type), Apaf-1−/−, or caspase-3−/− knockout mice were treated for 24 h with 2 μM STS (A and B), 100 μM etoposide, 150 μM cisplatin, 50 μM arsenite (B) and/or 50 μM Z-VAD.fmk (right panels only), followed by determination of the fall of the ΔΨm (measured as a reduction of the red fluorescence emitted by the ΔΨm-sensitive dye JC-1), mitochondrio-nuclear, and mitochondrio-cytosolic translocation of AIF or Cyt-c respectively (determined by immunostaining), proteolytic activation of caspase-3 (detected with an antibody specific for active caspase-3), or nuclear morphology (detected with Sytox-green). Individual cells demonstrated in A are shown after 8 h (wild-type) or 24 h (Apaf-1−/−, caspase-3−/−) of treatment with STS and are representative for the dominant phenotype. The chromatin condensation of wild-type cells cultured with STS was regarded as stage II of nuclear apoptosis, whereas Apaf-1−/− or caspase-3−/− cells manifest a stage I pattern of chromatin condensation. Kinetic analyses of apoptotic parameters induced by STS (▵), etoposide (□), cisplatin (▪), arsenite (○) and/or Z-VAD.fmk (right panels) are shown in B. This experiment has been repeated three times, yielding comparable results. Electron microscopy (C), large scale DNA fragmentation (D), and oligonucleosomal DNA fragmentation (E) of cells treated as in A are also shown.



The Apaf-1/Caspase/CAD Pathway and AIF Account for Two Different Phenotypes of Nuclear Apoptosis.
Wild-type, Apaf-1−/−, or caspase-3−/− cells were microinjected with Cyt-c, active caspase-3, CAD, or AIF. As expected 8 24, Cyt-c alone induced signs of apoptosis (ΔΨm loss, chromatin condensation) in wild-type cells, but not in Apaf-1−/− nor in caspase-3−/− cells (Fig. 2A and Fig. B). In contrast, microinjection of active caspase-3 provoked full-blown apoptosis (stage II) in all three cell types (Fig. 2 B). Similarly, AIF and CAD induced apoptosis in all cell types (Fig. 2A and Fig. B). AIF induced a peripheral type of chromatin condensation (similar to the caspase-independent stage I, Fig. 1 A), whereas CAD (and its activator caspase-3) provoked a more advanced pattern of nuclear compaction (similar to the caspase-dependent stage II, Fig. 1 A). The differential effect of AIF and CAD was confirmed in a cell-free system. When added to purified HeLa nuclei, AIF caused peripheral chromatin condensation (Fig. 3A and Fig. B), whereas CAD induced a much stronger type of chromatin compaction accompanied by a reduction in nuclear size (Fig. 3A and Fig. B). Moreover, AIF alone caused large scale DNA fragmentation (Fig. 3 D), yet was unable to provoke the “ladder type” oligonucleosomal chromatin digestion (Fig. 3 E). In contrast, CAD provoked the digestion of DNA in two steps, first into ∼50 kb (at low doses; Fig. 3 F) and then into mono- and oligomers of ∼200 bp (at high doses; Fig. 3 E). Moreover, CAD could act on AIF-pretreated nuclei (which lack oligonucleosomal DNA fragmentation, Fig. 3 E) to induce oligonucleosomal fragmentation (Fig. 3 G). In conclusion, AIF and CAD cause two morphologically and biochemically distinct types of nuclear apoptosis.
Figure 2.

Microinjection of apoptosis-regulatory proteins into Apaf-1−/− and caspase-3−/− cells. MEFs from control mice (wild-type), Apaf-1−/−, or caspase-3−/− knockout mice were microinjected with the indicated protein (5 μM AIF, 5μM AIFΔ1-351, 25 μM Cyt-c 1U/μl caspase-3, 250 nM ICAD/CAD protein, or 250 nM activated CAD), followed by staining with Hoechst 33342 (blue fluorescence) and the ΔΨm-sensitive dye CMXRos (red fluorescence). Representative phenotypes obtained 2 h after injection are shown in A. Mean values ± SD of three independent experiments are shown in B. Note that two stages (I and II) of nuclear chromatin condensation can be distinguished. Stage I resembles the nuclear apoptosis of Apaf-1 −/− cells (Fig. 1A and Fig. c), whereas stage II corresponds to that observed in apoptotic wild-type cells stimulated with STS (Fig. 1A and Fig. c).
Figure 3.

Apoptotic nuclear features induced by AIF and CAD in a cell-free system. HeLa nuclei were exposed to 200 nM AIF (120 min), 250 nM activated CAD (recombinant CAD/ICAD protein digested with caspase-3 inhibited or not with Ac-DEVD.fmk; 15 or 120 min), followed by staining with Hoechst 33342 (A) or electron microscopy (B). (C–E) In addition, nuclei were left untreated 1 or incubated for 120 min with: 200 nM AIF 2; AIF and 30 μM of PCMPS 3; AIF and 500 nM ICAD 4; caspase-3 5; 250 nM CAD–ICAD 6; 250 nM activated CAD 7; CAD–ICAD treated with 50 μM of Ac-DEVD.fmk before addition of caspase-3 8; CAD–ICAD treated with 50 μM of Ac-DEVD.fmk after activation of CAD by caspase-3 9; active CAD and ICAD 10; and active CAD and PCMPS. This was followed by staining with propidium iodide and flow cytometric determination of the frequency of hypoploid nuclei (C; X ± SD of four experiments), determination of large scale DNA fragmentation (D) and assessment of oligonucleosomal DNA fragmentation (E). (F) The kinetics of large scale DNA fragmentation induced by CAD (100 nM) was determined at different time points: nuclei untreated 1; incubated for 5 min 2; incubated for 15 min 3; incubated for 30 min 4; or incubated for 60 min 5. (G) Oligonucleosomal DNA fragmentation was assessed after exposure of control nuclei 1 to AIF alone (200 nM; 120 min; 2), CAD alone (250 nM; 120 min; 3), or after sequential exposure of nuclei, first to AIF, then to CAD 4.
CAD and AIF Act in Parallel Pathways of Nuclear Apoptosis.
As described above, AIF and CAD induced nuclear apoptosis independently from each other, both upon microinjection into intact cells (Fig. 2) and upon addition to purified nuclei (Fig. 3). ICAD inhibited CAD, yet did not antagonize AIF (Fig. 3C–E). Cytosolic extracts from wild-type MEFs undergoing apoptosis in response to STS, etoposide, or cisplatin were found to contain a biological activity which caused nuclear DNA loss in vitro upon addition to purified HeLa cells. Addition of ICAD failed to block this activity (Fig. 4A and Fig. B). Similarly, immunodepletion of AIF had no major inhibitory effect on such extracts (Fig. 4A and Fig. B). However, if AIF immunodepletion was combined with ICAD, apoptotic DNA loss was abolished (Fig. 4A and Fig. B). Cytosolic extracts from Apaf-1−/− and caspase-3−/− cells treated with different apoptosis inducers also contained an activity which induced nuclear apoptosis in the cell-free system (Fig. 4 B). In contrast to wild-type extracts, AIF depletion from Apaf-1−/− and caspase-3−/− extracts sufficed to inhibit the apoptosis-inducing activity, indicating that AIF is the principal factor responsible for nuclear apoptosis in such cells.
Figure 4.
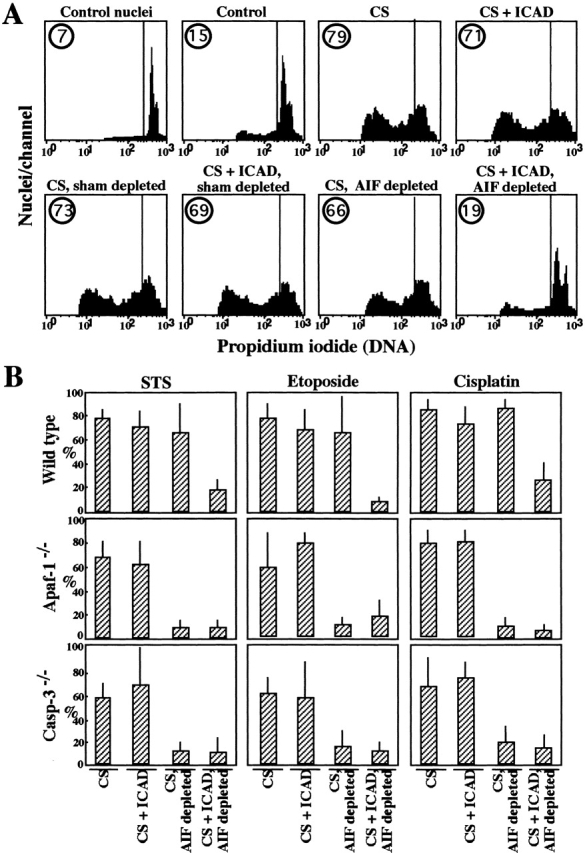
Inhibition of AIF and CAD reveals two parallel pathways of nuclear apoptosis. (A) HeLa nuclei were left untreated (control nuclei) or incubated for 2 h with cytosolic extracts obtained from untreated cells (Control) or cytosols (CS) from STS-treated wild-type MEFs, followed by staining with propidium iodide and flow cytometric quantitation of DNA content. Extracts were subjected to immunodepletion of AIF, sham immunodepletion, and/or addition of recombinant ICAD protein (500 nM). (B) Comparison of cytosols obtained from wild-type, Apaf-1−/−, and caspase-3−/− MEFs stimulated with three different apoptosis inducers (STS, etoposide, and cisplatin). Cytosols were evaluated for their capacity to induce nuclear DNA loss in purified HeLa nuclei (quantitated as in A) after depletion of AIF and/or addition of ICAD, as indicated. Cytosols from untreated control cells induced ≥10% of nuclear apoptosis. Results are means of triplicates (X ± SD) and are representative of three independent determinations.
Concluding Remarks.
Apoptosis-associated chromatin condensation and degradation may be expected to serve two purposes, namely to facilitate shrinkage (which in turn facilitates heterophagic removal of the apoptotic cells) and to prevent the DNA of the dead cell, which may include viral genomes or mutated genes including oncogenes, from being incorporated into adjacent cells. As shown here, at least two parallel and redundant pathways lead to nuclear apoptosis. One of these pathways involves caspases, ICAD, and CAD, and leads to oligonucleosomal DNA fragmentation and advanced chromatin condensation. The second, caspase-independent, pathway involves AIF and leads to large-scale DNA fragmentation and peripheral chromatin condensation (Fig. 1 Fig. 2 Fig. 3). These pathways may be activated in a sequential fashion, as the AIF-induced phenotype of nuclear apoptosis (stage I) normally precedes that induced by CAD (stage II; Fig. 1 and Fig. 5) and large scale DNA fragmentation precedes internucleosomal DNA cleavage in several models of apoptosis 15 25 26 27. Moreover, both pathways can act in a redundant fashion as suggested by the fact that, in a cell-free system involving cytoplasmic extract as apoptosis inducer, nuclear apoptosis is only prevented when both CAD and AIF are inhibited (Fig. 4 and Fig. 5). Recently, the existence of another chromatin condensation factor, Acinus, which requires activation by caspase-3 and another, unknown, protease has been reported 5. It may be speculated that Acinus is not fully activated in the cytosolic extract system used herein or, alternatively, that Acinus requires activation by AIF and/or is inhibited by ICAD.
Figure 5.

Scenario for the different phases of nuclear Apoptosis. AIF causes large scale chromatin fragmentation and a peripheral chromatin condensation (stage I). CAD causes a more advanced oligonucleosomal DNA fragmentation and DNA condensation (stage II).
In apparent contrast to the data reported here and in reference 4, a caspase-resistant mutant of ICAD completely prevents STS-induced DNA fragmentation in Jurkat cells, even at the level of large-scale DNA fragmentation 28. This may imply that the AIF pathway was not activated (or blocked?) in this cell type. The fact that the contribution of AIF (or other factors) to nuclear apoptosis varies is also suggested by variations in the observed effect of caspase inhibitors on apoptotic nuclear ultrastructure in different experimental systems, ranging from suppression of most signs of chromatin condensation 13 to no inhibition at all 10 11 12. AIF is expressed in an ubiquitous fashion 14, suggesting that factors other than AIF expression, e.g., endogenous AIF inhibitors or the absence of AIF targets, must explain these differences. Microinjection of an anti-AIF antibody prevents chromatin condensation induced by atractyloside or staurosporine 14, whereas depletion of AIF from cytosolic extracts (of wild-type cells) does not suffice to prevent chromatin condensation induced by such extracts (Fig. 4). This apparent paradox may reflect the fact that AIF is released from mitochondria before Cyt-c, correlating with stage I of apoptosis 15. Thus, when choosing a suitable time frame in short-term microinjection experiments, neutralization of AIF prevents the appearance of stage I of apoptosis. Moreover, AIF is a factor that causes the release of Cyt-c 14. Thus, AIF neutralization may be expected to retard the release of Cyt-c and the subsequent caspase/CAD activation that leads to stage II chromatin condensation.
Irrespective of these theoretical considerations, our data indicate the existence of at least two pathways leading to chromatin condensation and degradation during apoptosis. Why the process of apoptotic chromatin condensation is so complex and whether these pathways are connected at the molecular level by a common action on sessile nuclear proteins with proapoptotic potential remains an open question for future investigation.
Acknowledgments
We thank Anu Srinivasan (Idun Pharmaceuticals, San Diego, CA) for the CM1 caspase-3 antibody and Christine Schmitt for expert technical assistance.
L. Ravagnan and K.F. Ferri received Ph.D. fellowships from the French Ministry of Science & Technology; M. Loeffler received a postdoctoral fellowship from the Austrian Science Foundation; and P. Costantini received a fellowship from the Fondation pour la Recherche Medicale (FRM). This work has been supported by a special grant of the Ligue Nationale contre le Cancer, as well as by grants from Agence National de Recherche sur la SIDA, FRM (to G. Kroemer), Assistance Publique-Hôpitaux de Paris and Caisse Nationale Assurance Maladie (CANAM; contract 98006 to E. Daugas), and the Wellcome Trust (to W.C. Earnshaw).
Footnotes
S.A. Susin and E. Daugas contributed equally to this paper.
References
- Liu X., Zou H., Slaughter C., Wang X. DFF, a heterodimeric protein that functions downstream of caspase 3 to trigger DNA fragmentation during apoptosis. Cell. 1997;89:175–184. doi: 10.1016/s0092-8674(00)80197-x. [DOI] [PubMed] [Google Scholar]
- Enari M., Sakahira H., Yokoyoma H., Okawa K., Iwamtsu A., Nagata S. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature. 1998;391:43–50. doi: 10.1038/34112. [DOI] [PubMed] [Google Scholar]
- Sakahira H., Enari M., Nagata S. Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature. 1998;391:96–99. doi: 10.1038/34214. [DOI] [PubMed] [Google Scholar]
- Samejima K., Toné S., Kottke T.J., Enari M., Sakahira H., Cooke C.A., Durrieu F., Martins L.M., Nagata S., Kaufmann S.H., Earnshaw W.C. Transition from caspase-dependent to caspase-independent mechanisms at the onset of apoptotic execution. J. Cell Biol. 1998;143:225–239. doi: 10.1083/jcb.143.1.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahara S., Aoto M., Eguchi Y., Imamoto N., Yoneda Y., Tsujimoto Y. Acinus, a caspase-3 activated protein required for apoptosis chromatin condensation. Nature. 1999;401:168–171. doi: 10.1038/43678. [DOI] [PubMed] [Google Scholar]
- Zheng T.S., Schlosser S.G., Dao T., Hingorani R., Crispe N., Boyer J.L., Flavell R.A. Caspase-3 controls both cytoplasmic and nuclear events associated with Fas-mediated apoptosis in vivo. Proc. Natl. Acad. Sci. USA. 1998;95:13618–13623. doi: 10.1073/pnas.95.23.13618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo M., Hakem R., Soengas M.S., Ducan G.S., Shahinian A., Kägi D., Hakem A., McCurrach M., Khoo W., Kaufman S.A. Essential contribution of caspase 3/CPP32 to apoptosis and its associated nuclear changes. Genes Dev. 1998;12:806–819. doi: 10.1101/gad.12.6.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H., Kong Y.-Y., Yoshida R., Elia A.J., Hakem A., Hakem R., Penninger J.M., Mak T.W. Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell. 1998;94:739–750. doi: 10.1016/s0092-8674(00)81733-x. [DOI] [PubMed] [Google Scholar]
- Liu X.S., Li P., Widlack P., Zou H., Luo X., Garrard W.T., Wang X.D. The 40-kDa subunit of DNA fragmentation factor induces DNA fragmentation and chromatin condensation during apoptosis. Proc. Natl. Acad. Sci. USA. 1998;95:8461–8466. doi: 10.1073/pnas.95.15.8461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deas O., Dumont C., MacFarlane M., Rouleau M., Hebib C., Harper F., Hirsch F., Charpentier B., Cohen G.M., Senik A. Caspase-independent cell death induced by anti-CD2 or staurosporine in activated human peripheral T lymphocytes. J. Immunol. 1998;161:3375–3383. [PubMed] [Google Scholar]
- Berndt C., Möpps B., Angermüller S., Gierschik P., Krammer P.H. CXCR4 and CD4 mediate a rapid CD95-independent cell death in CD4+ cells. Proc. Natl. Acad. Sci. USA. 1998;95:12556–12561. doi: 10.1073/pnas.95.21.12556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quignon F., DeBels F., Koken M., Feunteun J., Ameisen J.C., de Thé H. PML induces a novel caspase-independent death process. Nat. Gen. 1998;20:259–265. doi: 10.1038/3068. [DOI] [PubMed] [Google Scholar]
- Hirsch T., Marchetti P., Susin S.A., Dallaporta B., Zamzami N., Marzo I., Geuskens M., Kroemer G. The apoptosis-necrosis paradox. Apoptogenic proteases activated after mitochondrial permeability transition determine the mode of cell death. Oncogene. 1997;15:1573–1582. doi: 10.1038/sj.onc.1201324. [DOI] [PubMed] [Google Scholar]
- Susin S.A., Lorenzo H.K., Zamzami N., Marzo I., Snow B.E., Brothers G.M., Mangion J., Jacotot E., Costantini P., Loeffler M. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- Daugas E., Susin S.A., Zamzami N., Ferri K., Irinopoulos T., Larochette N., Prevost M.C., Leber B., Andrews D., Penninger J., Kroemer G. Mitochondrio-nuclear redistribution of AIF in apoptosis and necrosis. FASEB (Fed. Am. Soc. Exp. Biol.) J. 2000;14:729–739. [PubMed] [Google Scholar]
- Marzo I., Brenner C., Zamzami N., Jürgensmeier J., Susin S.A., Vieira H.L.A., Prévost M.-C., Xie Z., Mutsiyama S., Reed J.C., Kroemer G. Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science. 1998;281:2027–2031. doi: 10.1126/science.281.5385.2027. [DOI] [PubMed] [Google Scholar]
- Srinivasula S.M., Fernandes Alnemri T., Zangrilli J., Robertson N., Armstrong R.C., Wang L.J., Trapani J.A., Tomaselli K.J., Litwack G., Alnemri E.S. The Ced-3/interleukin 1 beta converting enzyme-like homolog Mch6 and the lamin-cleaving enzyme Mch2 alpha are substrates for the apoptotic mediator CPP32. J. Biol. Chem. 1996;271:27099–27106. doi: 10.1074/jbc.271.43.27099. [DOI] [PubMed] [Google Scholar]
- Srinivasan A., Roth K.A., Sayer R.O., Shindler K.S., Wong A.N., Fritz L.C., Tomaselli K.J. In situ immunodetection of activated caspase-3 in apoptotic neurons in the developing nervous system. Cell Death Differ. 1998;5:1004–1016. doi: 10.1038/sj.cdd.4400449. [DOI] [PubMed] [Google Scholar]
- Kawabe Y., Ochi A. Programmed cell death and extrathymic reduction of Vβ8+ CD4+ T cells in mice tolerant to Staphylococcus aureus enterotoxin B. Nature. 1991;349:245–248. doi: 10.1038/349245a0. [DOI] [PubMed] [Google Scholar]
- Brown D.G., Sun X.M., Cohen G.M. Dexamethasone-induced apoptosis involves cleavage of DNA to large fragments prior to internucleosomal fragmentation. J. Biol. Chem. 1993;268:3037–3039. [PubMed] [Google Scholar]
- Susin S.A., Lorenzo H.K., Zamzami N., Marzo I., Larochette N., Alzari P.M., Kroemer G. Mitochondrial release of caspases-2 and -9 during the apoptotic process. J. Exp. Med. 1999;189:381–394. doi: 10.1084/jem.189.2.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susin S.A., Zamzami N., Castedo M., Hirsch T., Marchetti P., Macho A., Daugas E., Geuskens M., Kroemer G. Bcl-2 inhibits the mitochondrial release of an apoptogenic protease. J. Exp. Med. 1996;184:1331–1342. doi: 10.1084/jem.184.4.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susin S.A., Zamzami N., Castedo M., Daugas E., Wang H.-G., Geley S., Fassy F., Reed J., Kroemer G. The central executioner of apoptosis. Multiple links between protease activation and mitochondria in Fas/Apo-1/CD95- and ceramide-induced apoptosis. J. Exp. Med. 1997;186:25–37. doi: 10.1084/jem.186.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H., Henzel W.J., Liu X., Lutschg A., Wang X.D. Apaf-1, a human protein homologous to C. elegans Ced-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90:405–413. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]
- Oberhammer F., Wilson J.W., Dive C., Morris I.D., Hickman J.A., Wakeling A.E., Walker P.R., Sikorska M. Apoptotic death in epithelial cellscleavage of DNA to 300 and/or 50 kb fragments prior to or in the absence of internucleosomal fragmentation. EMBO (Eur. Mol. Biol. Organ) J. 1993;12:3679–3684. doi: 10.1002/j.1460-2075.1993.tb06042.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhivotovsky B., Wade D., Gahm A., Orrrenius S., Nicotera P. Formation of 50 kbp chromatin fragments in isolated liver nuclei is mediated by protease and endonuclease activation. FEBS (Fed. Eur. Biochem.) Let. 1994;351:150–154. doi: 10.1016/0014-5793(94)00827-2. [DOI] [PubMed] [Google Scholar]
- Lagarkova M.A., Iarvaia O.V., Razin S.V. Large-scale fragmentation of mammalian DNA in the course of apoptosis proceeds via excision of chromosomal DNA loops and their oligomers. J. Biol. Chem. 1995;270:20239–20241. doi: 10.1074/jbc.270.35.20239. [DOI] [PubMed] [Google Scholar]
- Sakahira H., Enari M., Ohsawa Y., Uchiyama Y., Nagata S. Apoptotic nuclear morphological change without DNA fragmentation. Curr. Biol. 1999;9:543–546. doi: 10.1016/s0960-9822(99)80240-1. [DOI] [PubMed] [Google Scholar]