

The diversion of most cellular energy to fuelling actomyosin during exercise comes at a cost – a ‘switch off’ of protein synthesis during and immediately after contractile activity. Thereafter as the old adage ‘what doesn't kill you makes you stronger’ suggests, the period of rest and feeding after exercise helps us to bounce back stronger. Nevertheless we are still remarkably ignorant of what actually controls protein maintenance during and immediately after exercise. Only relatively recently have workers measured both protein synthesis and cellular signalling activities in exercise-related studies and started to tease out the differential effects of mode and intensity of exercise (Gautsch et al. 1998; Atherton et al. 2005), especially in relation to alterations in protein synthesis and the processes controlling the initiation and elongation phases of translation. Although there have been a number of observations of changes in phosphorylation state and (presumed activity) in important candidates for inclusion in the control network the field has been bedevilled by a plethora of molecular changes which has obscured a clear view of the actual important behaviours in altering the processes controlling protein turnover during and after exercise.
All the following signalling enzymes are capable of modulating the synthetic apparatus and their activation has been observed as a result of many kinds of contractile activity in a variety of models: mitogen activated protein kinase pathway (MAPK) family members (e.g. ERK1/2, p38) (Williamson et al. 2003); proteins of the canonical insulin receptor signalling pathway (e.g. Akt, mTOR) (Atherton et al. 2005); contraction-induced Ca2+-dependent pathways (e.g. PKC, CamK) (Rose et al. 2005) and AMPK (Frosig et al. 2004) to name but a few. What is very confusing is that many proteins of these networks are capable of positive and negative cross-talk, so that activation of a specific signalling element does not necessarily relate to an observed effect on initiation of protein synthesis or mRNA translation – there may be redundancy and negative feedback as well as amplification. So which are the key players?
Answering this to create a definitive scheme of exercise-induced signalling has been hampered by fragmented approaches including different exercise modes (e.g. resistance versus endurance); in vivo or in vitro animal models; training status and the choice of mixed signalling targets and phosphorylation sites.
In this issue of The Journal of Physiology, Williamson et al. (2006) provide a more comprehensive account than previously available of translational signalling during running exercise. They describe alterations in the phosphorylation state of a range of proteins from pathways involving MAPK, the insulin receptor and AMPK. Among the findings is the observation (as made previously by numerous workers) that phosphorylation of MAPK proteins MEK1/2, ERK1/2 and p90RSK1 was increased, which theoretically should act to increase mTOR signalling and translational rates concomitantly via both ERK1/2 and p90RSK1 induced phosphorylation of TSC1/2 and ERK1/2 of eIF4E. The problem is that there is apparently reduced translational signalling during exercise, including Ca2+–calmodulin activation of eEF2 kinase (Rose et al. 2005). However, Williamson et al. (2006) also provide evidence that the potentially positive effect of the mitogen pathway elements is overridden by the negative effect of AMPK on TSC1/2–mTOR signalling. Indeed cAMP, upstream PKA activity and AMPK phosphorylation itself was increased at all times during exercise coincident with decreased polysome aggregation, a semi-quantitative index of translational initiation. They also noted differences in the status of association between raptor–mTOR and TSC1–TSC2, both of which may negatively modulate the ability of these complexes to transduce signals. Precisely how well the association such as that of mTOR–raptor matches potential for kinase activity in response to exercise remains to be seen. In light of this it would have been nice if the authors were to have measured mTOR activity and phosphorylation at Thr2446 since this might have clarified non-TSC2-dependent AMPK-induced repression.
Nevertheless, as well as helping to answer some puzzles, the work suggests new puzzles yet to be solved. For example, because usually, mTOR-dependent signalling co-regulates p70 and 4E-BP1, why, if mTOR signalling is inhibited is there no subsequent dephosphorylation of p70 as for 4E-BP1? The authors failed to see any Thr389 phosphorylation or significant p70 hypophosphorylation during exercise, but if they had measured other sites such as Thr421/Ser424, this may have provided an explanation because p70 may also be phosphorylated by PKC and MAPK-dependent pathways. Furthermore if most translational regulators are switched off during exercise then why are isolated examples such as rpS6 still activated? May they aid selective translation, have unknown functions, or are they simply not absolutely necessary? Furthermore, what about elongation? Interestingly the authors reported increased p90RSK1 phosphorylation which has been shown to inactivate eEF2 kinase (Wang et al. 2001) theoretically providing a stimulatory effect on elongation; therefore perhaps activation of eEF2 kinase through p70 deactivation, AMPK phosphorylation or simply by non-covalent Ca2+–calmodulin-dependent mechanisms overrides this effect. Of course for those of us greedy to understand more about the nature of the exercise-induced effects we want to know more – what about protein breakdown; the relationship with Ca2+-dependent mechanisms (Rose et al. 2005), the kinetics of post-exercise changes and as always, does this mechanism occur in people, and what are the implications for exercise training for runners? But it would be churlish to deny the usefulness of this work simply because it does not provide all the answers. The approach and novelty are salutary.
Figure 1. Scheme showing possible events controlling the inhibition of muscle protein synthesis during exercise.
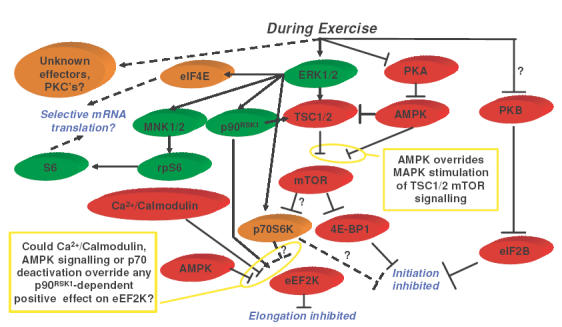
Arrow: activation status, positive influence on translation (influenced through proteins in green). Perpendicular line: activation status, negative influence on translation (influenced through proteins in red). Dashed lines, effects unclear (proteins in light orange). ?, contrasting data from other models, thus effect unclear.
References
- Atherton PJ, Babraj J, Smith K, Singh J, Rennie MJ, Wackerhage H. Faseb J. 2005;19:786–788. doi: 10.1096/fj.04-2179fje. [DOI] [PubMed] [Google Scholar]
- Frosig C, Jorgensen SB, Hardie DG, Richter EA, Wojtaszewski JF. Am J Physiol Endocrinol Metab. 2004;286:E411–E417. doi: 10.1152/ajpendo.00317.2003. [DOI] [PubMed] [Google Scholar]
- Gautsch TA, Anthony JC, Kimball SR, Paul GL, Layman DK, Jefferson LS. Am J Physiol. 1998;274:C406–C414. doi: 10.1152/ajpcell.1998.274.2.C406. [DOI] [PubMed] [Google Scholar]
- Rose AJ, Broholm C, Kiillerich K, Finn SG, Proud CG, Rider MH, Richter EA, Kiens B. J Physiol. 2005;569:223–228. doi: 10.1113/jphysiol.2005.097154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Li W, Williams M, Terada N, Alessi DR, Proud CG. EMBO J. 2001;20:4370–4379. doi: 10.1093/emboj/20.16.4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson D, Gallagher P, Harber M, Hollon C, Trappe S. J Physiol. 2003;547:977–987. doi: 10.1113/jphysiol.2002.036673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson D, Kubica N, Kimball SR, Jefferson LS. J Physiol. 2006;573:497–510. doi: 10.1113/jphysiol.2005.103481. [DOI] [PMC free article] [PubMed] [Google Scholar]