

Abstract
Type I interferon (IFN) is synthesized by most nucleated cells following viral infection. Robust IFN production in cell culture requires positive feedback expression of inducible signaling components, such as the transcription factor IRF7. However, the role of positive feedback and IRF7 in vivo may be more complex. We found that IFN produced locally in the respiratory tract of influenza virus-infected mice displayed characteristics of positive feedback, including Stat1-dependent induction of IRF7 and IFN gene expression. IRF7 expression was similarly stimulus-dependent in most tissues. However, lymphoid tissue constitutively expressed high levels of IRF7 in the absence of induction or positive feedback, and this expression was largely confined to plasmacytoid dendritic cells (DC). These cells rapidly produced large quantities of multiple IFNα species following viral infection without positive feedback, whereas other hematopoietic cells, including other DC subtypes, expressed little IRF7 and were poor IFN producers in the absence of positive feedback. These data reveal a dual mechanism for the regulation of IFN production by differential expression of IRF7, involving positive feedback at local sites of infection combined with robust systemic production by IRF7-expressing plasmacytoid DC.
Type I IFN (IFNα/β)1 is produced by virally infected cells. These cytokines play important roles in the response to both local and systemic infections by signaling through the type I IFN receptor complex (IFNAR1 and IFNAR2). Type I IFN induces numerous genes known generally as IFN-stimulated genes (ISG) that encode proteins involved in innate and adaptive antiviral immune responses (reviewed in Ref. 1). The type I IFN receptor is associated with the Janus protein tyrosine kinases JAK1 and TYK2 that target the STAT transcription factors STAT1 and STAT2 for phosphorylation. Phosphorylation-dependent dimerization of STAT1 and STAT2 and interaction with the DNA-binding protein IRF9 lead to assembly of the transactivation complex ISGF3 responsible for induction of immediate-early target genes. Among the immediate-early class of IFN-stimulated genes is the transcription factor IRF7. IRF7 along with the related protein IRF3 controls the production of type IIFN in most infected cells following phosphorylation-dependent activation (reviewed in Ref. 2).
Current models for IFNα induction following viral infection invoke activation of cellular protein serine kinases by viral signals, leading to a kinase cascade culminating in phosphorylation of IRF3 (3). Activated IRF3 is sufficient to induce expression of one subtype of IFNα (IFNα4 in mice, IFNA1 in human) and collaborates with activated AP1 and NFκB transcription factors to induce IFNβ. However, transcription of additional IFNα subtype genes requires IRF7, which must first be synthesized in response to this early limited IFN production, thus establishing a positive feedback loop (4, 5). The kinase complex implicated in both IRF3 and IRF7 phosphorylation consists of the enzymes TBK1 and IκB kinase ε (6, 7) and is activated by cellular intermediaries that detect viral infection, such as the RNA helicases RIG-I and MDA-5 (8, 9). Like IRF7, IκB kinase ε, RIG-I, and MDA-5 are also IFN-inducible proteins, as are the ISGF3 components STAT1 and IRF9. Although ISGF3 is not directly involved in IFN gene transcription, it is required for the induction of all these IFN-responsive components of the positive feedback amplification loop (10, 11). Therefore, mice lacking STAT1 cannot mount positive feedback and are limited to production of the immediate-early IFN subtypes α4 and β when challenged with a respiratory infection (4). Impaired IFN production in concert with impaired IFN responsiveness contributes to the exquisite sensitivity of STAT1-deficient mice to viral infection (10–13).
The importance of feedback and the role of IRF7 were investigated by Kalinke and colleagues (14), who studied IFNα production by splenocytes following infection with vesicular stomatitis virus or UV-irradiated Herpes simplex virus. They observed that at early time points (<6 h post-infection), marginal zone splenocytes produced similar quantities of IFN irrespective of whether the mice expressed IFN receptors or not. Coupled with an inability to detect significant levels of IRF7 mRNA in uninfected splenocytes, they concluded that early IFN production in vivo did not require IFN feedback amplification and questioned the role of IRF7 in this process (14).
However, understanding induction of IFN in vivo and the requirement for positive feedback may be complicated by cell type-specific differences. For instance, a blood cell has been identified, known as the interferon-producing cell (15), which is a consummate producer of IFN and appears to be a particular subtype of dendritic cell (DC), known as the plasmacytoid DC (pDC) (16, 17). This cell type appears to have distinct mechanisms for producing IFN (18, 19), at least in response to inducers that signal through Toll-like receptors (TLRs). Adding complexity to the issue, the exact cell type that makes type I IFN may depend on the nature of the infectious agent. Biron and colleagues (20) showed that both murine cytomegalovirus and lymphocytic choriomeningitis virus infections resulted in IFN production, but IFN induced by the former originated from splenic pDC, whereas IFN in response to the lymphocytic choriomeningitis virus came from a non-splenic non-pDC source. Moreover, further work suggested that the infected cell and the IFN-producing cell may not necessarily be the same (21). Interestingly, non-replicating inducers (such as soluble toxo-plasma antigen) provoked cytokine production from distinct subsets of DC when compared with infections with live replicating microbes (21). In an attempt to resolve these divergent results, we have previously hypothesized that the route of infection may influence the responsive cell type (22). Systemic infections (e.g. intravenous) may lead to efficient activation of splenic pDC, whereas peripheral infections (e.g. respiratory) may only target localized responsive cell types.
To further investigate this hypothesis, we have used in vivo and in vitro infection models and investigated the role of IRF7 induction and positive feedback in IFN production in response to negative-strand RNA viruses. We found that IFN production correlated with IRF7 production in infected lungs, which was drastically impaired in STAT1-deficient mice, consistent with a requirement for positive feedback. However, although most mouse tissues examined displayed induction-dependent production of IRF7, pDC in the spleen or differentiated in vitro expressed high levels of constitutive IRF7 protein and responded rapidly to virus infection by robust production of multiple IFNα species. Thus, our results pinpoint differential regulation of IRF7 expression as the underlying mechanism of positive IFN feedback regulation in peripheral tissues and as an important element of cell type-specific differences in IFN production.
MATERIALS AND METHODS
Mice
Mice deficient for STAT1 (stat1−/−) or for Rag1 (rag1−/−) and wild type controls on the same inbred background (BALB/c and C57BL6/J) were bred under specific pathogen-free conditions, in accordance with approved protocols at New York University School of Medicine. All mice were 8–12 weeks of age. In some experiments, IFN was induced in vivo by intraperitoneal injection of 100 μg of poly(I-C) per mouse 1 day before collecting organs for protein extraction. Individual whole organ protein extracts were obtained by crushing tissue between frosted glass slides and lysing the resulting released cells in immunoprecipitation lysis buffer (1% Triton X-100, 300 mm NaCl, 50 mm HEPES pH 7.6, 1 mm Na3VO4, 1 mm dithiothreitol, 1 mm phenylmethylsulfonyl fluoride, and 5 μg/ml aprotinin and leupeptin).
Enrichment of Splenic Subsets Using Magnetic Beads
Isolated spleens were disrupted by digestion with Liberase CI (1 ml/spleen of 1.67 Wunsch units/ml; Roche Applied Science) and DNase I (0.2 mg/ml; Roche Applied Science) for 30 min at 37 °C. Single cell suspensions were passed through a cell strainer (70 μm), and red blood cells were lysed using red blood cell lysis buffer (155 mm ammonium chloride, 0.1 mm EDTA, 12 mm sodium bicarbonate). Cell subtype enrichment involved anti-CD11c- and anti-CD11b-coupled magnetic beads and positive selection column MS+, according to the manufacturer’s instructions (MACS, Miltenyi Biotec). Positive MACS bead selections typically yielded cells that were greater than 90% pure after one round of selection, as judged by flow cytometry.
Flow Cytometric Analyses
Cell surface staining was carried out in phosphate-buffered saline containing 0.5 mm EDTA unless otherwise stated. Cells were incubated with the following fluorochrome-conjugated monoclonal antibodies: CD11b-fluorescein isothiocyanate, CD11c-phycoerythrin, and IgM-fluorescein isothiocyanate (BD Biosciences). Cells were washed and fixed in 1% paraformaldehyde before analysis.
Hematology
Splenic cell preparations or in vitro differentiated bone marrow (BM) were prepared by Cytospin centrifugation and stained with Wright-Giemsa stain.
Cell Isolation and Culture
BM cells were flushed from femurs and tibiae with RPMI 1640 medium with 25 mm Hepes. Red blood cells were lysed, and nucleated cells were plated in the appropriate medium. FLT3L-differentiated cells were plated at 106/ml (day 0) and expanded in RPMI 1640 containing 1% syngeneic normal mouse serum, sodium pyruvate (1 mm), HEPES (25 mm), β-mercaptoethanol (2 mm), and antibiotics (penicillin-streptomycin; Cellgro), and were grown for 8 days in 24-well plates in the presence of 25 ng/ml FLT3L (Peprotech) and 10 ng/ml stem cell factor (Peprotech) with 80% of the medium replenished on day 5. GM-CSF-differentiated DC cultures were generated essentially as described by Inaba et al. (23). In brief, BM cells were plated at 700,000 cells/ml (day 0) in RPMI 1640 containing 10% fetal calf serum, HEPES (25 mm), sodium pyruvate (1 mm), and antibiotics (penicillin-streptomycin; Cellgro) and supplemented with 40 ng/ml GM-CSF (Peprotech). On day 2, 70% of the medium was replenished. On day 3, non-adherent cells were removed, retaining the loosely adherent DC progenitor cells. The resulting differentiated and expanded cells were collected on day 5. Other cytokines used in some cultures were interleukin-3 (10 ng/ml; Peptrotech) and IFNα/β (500 units/ml; Access Biomedical).
Virus Infections
Mice were infected with influenza A virus (WSN strain) by inhalation, as described previously (12). In brief, 105 plaque-forming units in 50 μl of phosphate-buffered saline were instilled intranasally into each anesthetized wild type or STAT1 mutant mouse, and lungs of infected mice were harvested 1–3 d later. RNA was isolated from disrupted lungs by the TRIzol method, according to the manufacturer’s instructions (Invitrogen).
In vitro differentiated BM-derived cells (1 × 106) were cultured in 50 μl of RPMI 1640 containing 25 mm Hepes and 0.1% bovine serum albumin, either in the presence or in the absence of 50 μl (128 hemaglutination units/ml) of Newcastle disease virus (NDV). After 1 h at 37 °C, cells were plated in medium supplemented with 10% fetal calf serum for 4–12 h, as indicated, and cells were lysed in immunoprecipitation lysis buffer. For RNA analysis, 2 × 106 BM-derived cells were used for each infection, and medium, virus, and reagents were scaled up appropriately.
Antibodies
Immunoblotting was performed using the following polyclonal antibodies: anti-IRF7, anti-IRF3, anti-IRF4, and anti-IRF8 (Zymed Laboratories Inc.) and monoclonal α-tubulin (Sigma).
Quantitative Real-time RT-PCR Analysis
RNA was extracted from at least 2 × 106 BM-derived cells using the RNeasy Minikit (Qiagen). Reverse transcription was performed using 3 μg of total RNA, and the resulting cDNA was used for real-time PCR analysis, using gene-specific primer pairs in the presence of Sybr® Green dye (Molecular Probes, Eugene, OR). Each target transcript was quantified by comparison with a standard curve generated by serial dilution of an abundant sample following normalization to the amount of glyceraldehyde-3-phosphate dehydrogenase or ribosomal protein L32 mRNA. Sequences of real-time PCR primers are available upon request.
Gene Transduction
STAT1-deficient fibroblasts were reconstituted with IRF7 by recombinant retroviral infection, which was performed using Phoenix cells, following the Nolan laboratory protocol. In brief, Phoenix cells, which are 293T cells stably expressing ecotropic packaging proteins, were transfected with retroviral constructs (green fluorescent protein or empty vector retroviruses as controls) in the presence of 25 μm chloroquine using a standard calcium phosphate protocol. 48 h later, supernatants were collected, spun, and used directly for infection of 3T3 cells preplated a day earlier at 1 × 105/6 wells in the presence of 40 μg/ml polybrene.
RESULTS
Effective IFN Expression in Lungs following Infection Requires Intact IFN Signaling
An essential role for type I IFN in resolving respiratory infections and coordinating an appropriate T cell response has been documented previously (12, 13). Importantly, those studies revealed a role for IFN locally at the site of infection to orchestrate the inflammatory response as well as systemically to limit the spread of virus to peripheral tissues. In contrast to wild type animals, IFN-unresponsive mice (STAT1- or IFNAR-deficient) failed to contain influenza virus within the respiratory tract or establish an effective Th1-biased immune response. We investigated whether impaired IFN production at the site of infection due to the absence of positive feedback contributed to the phenotype of STAT1-deficient mice.
Induction of IFN was compared in wild type and STAT1 mutant mice following intranasal influenza viral infection in vivo. Although IFN was readily detected in infected lungs from wild type mice, IFN production was markedly impaired in STAT1-null lungs when compared with the wild type lungs (Fig. 1A, black squares). Additionally, the mutant lungs failed to induce the accumulation of IRF7 message to levels equivalent to wild type controls (Fig. 1A, open triangles). Thus, the inability to support positive feedback in vivo via induction of IRF7 within the respiratory epithelium likely contributed to the failure of STAT1 mutant mice to mount an effective IFN response in response to respiratory infection.
Fig. 1. Positive feedback-dependent induction of IRF7 and IFNα in vivo.

A, IRF7 and IFNα (all subtypes) levels were measured in lungs of wild type (WT) and stat1−/− mice (BALB/c), intranasally infected with influenza virus, by quantitative real-time RT-PCR. The data represent mean ± S.E. of three experiments. B, profile of IRF7 and IRF3 protein expression in tissues 1 day after intraperitoneal injection of poly(I-C) in wild type mice (C57BL6/J). Liv., liver; Thy., thymus; Spl., spleen; Tes., testes; MW, molecular masses. C, IRF7 protein expression levels in spleen and thymus of wild type and stat1−/− mice (C57BL6/J). ko, knock-out. D, comparison of IRF7 protein expression in bone marrow and spleen from wild type and rag1−/− mice (BALB/c). The data are representative of three (B and C) and two experiments (D), respectively. n.s., nonspecific.
IRF7 Protein Is Inducible in Most Tissues but Constitutively Expressed in Spleen
IRF7 was initially identified as an IFN-inducible transcription factor in hematopoietic cells (24). Given its induction in response to virus infection in lung, we examined its expression in multiple organs. To mimic systemic viral infection, mice were intraperitoneally injected with poly(I-C), a well characterized type I IFN inducer (25), and 1 day after poly(I-C) injection, selected organs were examined for IRF protein expression. Abundance of IRF7 protein increased in response to poly(I-C) injection in most tissues (Fig. 1B).
Interestingly, IRF7 protein was expressed at relatively high levels in thymus and spleen, even in the absence of poly(I-C) treatment. In contrast, IRF3 was expressed in all tissues examined, and its expression was not affected by IFN induction (Fig. 1B, lower panel). Similar results were obtained after infection of mice with negative-strand RNA viruses (data not shown). Thus, like lung, most peripheral tissues required induction to accumulate significant levels of IRF7 protein expression and would likely exhibit positive feedback-dependent production of IFN following viral infection. However, constitutive IRF7 accumulation in lymphoid organs indicated the possibility of a distinct type of response to infection in these tissues.
IFN Feedback Does Not Account for IRF7 Protein Levels in the Spleen
A possible explanation for elevated constitutive levels of IRF7 in spleen and thymus could be the low levels of type I IFN observed in lymphoid organs even in the absence of viral infection (26–28). To examine this possibility, IRF7 protein expression was quantified in multiple organs, including spleens and thymi of stat1−/− mice that are incapable of responding to IFN. Stat1−/− mice expressed levels of IRF7 in spleen and thymus comparable with wild type mice, whereas in other tissues, such as lung, heart, kidney, and skeletal muscle, IRF7 accumulation strictly required STAT1-dependent induction (Fig. 1C and data not shown). Therefore, IFN feedback does not appear to be required for abundant constitutive IRF7 protein expression in lymphoid organs.
Mature T Cells and B Cells Are Not Required for Splenic IRF7 Expression
The mouse spleen is composed largely of B and T cells, together constituting ~90% of splenic cellularity. To determine whether B and T cells were the origin of the abundant resting IRF7 levels observed in spleen, mice deficient for Rag1 protein and therefore lacking mature B and T cells were examined (Fig. 1D). Rag1−/− mouse spleen expressed equivalent levels of IRF7 protein when compared with wild type spleen, indicating that mature B and T cells were not the exclusive source for splenic IRF7 expression.
IRF7 protein expression was also directly measured in lymphocytes. T and B cells from wild type spleens were purified by magnetic bead selection using anti-CD3 and anti-CD19 antibodies, respectively. Little IRF7 protein was detected in lymphocytes not treated with IFN (data not shown), suggesting that splenic IRF7 expression was localized to a non-T non-B splenocyte population.
Kinetics of IFN Production and IRF7 Induction in Fibroblasts and Splenocytes
We examined how constitutive expression of IRF7 in lymphoid tissue impacted the kinetics of IFN production following virus infection. Comparing fibroblasts (inducible IRF7) with splenocytes (constitutive IRF7), we measured the rate of IFN induction and the role of positive feedback in response to infection. As shown in Fig. 2A, IRF7 mRNA was expressed at least 3 orders of magnitude higher in uninfected splenocytes (triangles) when compared with uninfected fibroblasts (squares). The presence or absence of STAT1 did not significantly affect constitutive IRF7 expression in spleen (compare open and closed symbols), confirming the lack of a role for constitutive IFN in maintaining high basal IRF7 expression in spleen. IRF7 levels were further induced following viral infection, but to a significant extent only in wild type cells, suggesting that positive feedback can further augment IRF7 production in response to autocrine IFN. As shown previously, IRF7 was also highly inducible in wild type fibroblasts, reaching levels equivalent to induced splenocytes but only at late times post-infection (p.i.). However, IRF7 induction in STAT1-deficient cells was significantly impaired.
Fig. 2. High constitutive expression of IRF7 in splenocytes correlates with accelerated and enhanced induction of IFNα.

A, IRF7 mRNA levels expressed at indicated times p.i. with NDV in spleens and fibroblasts from wild type or stat1−/− mice (C57BL6/J), as measured by quantitative real-time RT-PCR. B, IFNα mRNA levels (all subtypes) corresponding to the samples analyzed in A. WT, wild type. C, IFNα (non-α4 subtype) mRNA levels from NDV-infected wild type and stat1−/− spleens. Expression levels were normalized to internal controls (ribosomal protein L32 mRNA). The data are representative of four experiments.
Expression of high levels of constitutive IRF7 protein in wild type splenocytes correlated with accelerated and enhanced induction of type I IFN following virus infection (Fig. 2B and C). IFN levels produced by infected splenocytes at 4 h p.i. were comparable with those induced in infected wild type fibroblasts at 8–12 h p.i., subsequent to IRF7 induction. At 4 h p.i., IFN production by STAT1 mutant splenocytes was equivalent to that produced by wild type cells, which was consistent with their equivalent IRF7 levels. Greater IFN production was observed in wild type splenocytes at later time points, reflecting the induction of IRF7 (Fig. 2A), presumably in response to autocrine IFN. Little IFN was produced by STAT1-deficient fibroblasts, the IRF7 levels of which started out and remained low throughout the course of the infection.
Expression of IFNα subtypes other than IFNα4 is completely dependent on the presence of IRF7 and therefore on IFN sensitivity, at least in fibroblasts (4). A similar correlation with the presence of IRF7 was observed in splenocytes (Fig. 2C). The non-α4 IFN subset was induced in both wild type and STAT1-deficient splenocytes that expressed IRF7. Equal levels of IFN induction were observed at 4 h, although wild type cells expressed higher levels at later time points, subsequent to further induction of IRF7. Thus, expression of IFNα was dependent on IRF7 in both fibroblasts and splenocytes; however, the higher initial set point of IRF7 abundance in splenocytes allowed robust IFN production prior to feedback induction of IRF7 following autocrine IFN stimulation.
A DC Subset Is the Source of Abundant IRF7 in Splenocytes
To identify the cell type that accounted for high levels of IRF7 expression in spleen, individual non-B and non-T splenic cell populations were isolated and assayed for IRF7 expression. Splenic CD11c+ DC expressed relatively high levels of IRF7 protein, as well as moderately elevated levels of IRF8 (Fig. 3A, left panel). In contrast, other IRF proteins, such as IRF3, were expressed equally by both CD11c+ and CD11c− cell populations.
Fig. 3. IRF7 protein is highly expressed in a CD11b−CD11c+ DC subset.

A, splenocytes from wild type C57BL6/J mice were positively selected with MACS beads coupled to an anti-CD11c antibody, and the corresponding levels of IRF7, IRF3, IRF8, and IRF4 proteins were assayed (left). Similarly, splenocytes were positively selected sequentially with MACS beads coupled to anti-CD11b followed by anti-CD11c antibodies, and the corresponding levels of IRF7, IRF3, IRF8 and IRF4 proteins were assayed (right). Tub, tubulin. B, upper panels, naive BM from wild type C57BL6/J mice was cultured in vitro with combinations of FLT3L (25 ng/ml), interleukin-3 ((IL3) 10 ng/ml), IFN (500 units/ml), and GM-CSF (40 ng/ml), as indicated, to expand different DC subsets, and IRF7 and IRF3 protein levels were assessed. B, lower panels, IRF7 and IRF3 protein levels were measured in NDV-infected BM-derived DC generated using GM-CSF and FLT3L, as indicated. Protein extracts from equal numbers of cells (A, 1 × 106; B, 5 × 105) were loaded per lane. Mock, mock-infected. C, representative morphologies of cells expanded in vitro from BM in response to GM-CSF (panel a), FLT3L (panel b), or interleukin-3 (panel c) were determined by Giemsa staining of cytospin preparations. D, pDC sorted from spleen by FACS were examined for IRF expression by immunoblotting and compared with whole spleen and BM-derived mDC. Extract from 1 × 10 6 cells was loaded per lane. n.s., nonspecific.
To further characterize the IRF7-expressing splenocyte population, subset enrichment was carried out by sequential positive selection. CD11b+ cells were first selected to isolate CD11b+CD11c+ DC along with CD11b+ splenic macrophages. Subsequently, CD11c+ cells were selected from the CD11b− population, to isolate the CD11b−CD11c+ DC subset. The remaining double negative cells after CD11c isolation were also examined. FACS staining confirmed the effectiveness of the selections through the expression of CD11b and CD11c markers on the appropriate cell populations (data not shown). As shown in Fig. 3A (right panel), CD11b−CD11c+ DC expressed greater levels of IRF7 protein when compared with CD11b− or double negative cell populations. Again, IRF8 was also enriched in CD11b− CD11c+ cells, whereas other IRF proteins were expressed at similar levels in all subsets. Thus, IRF7 is expressed at highest levels in CD11b−CD11c+ cells, suggesting that it is highly expressed in a DC subset.
FLT3L-differentiated Bone Marrow Generates IRF7-expressing DC
Dendritic cells can be broadly divided into distinct subtypes, including myeloid DC (mDC) and plasmacytoid DC (pDC). Similar cells can be differentiated and expanded in vitro from BM progenitors. Conventional mDC expand in the presence of GM-CSF, whereas pDC progenitors respond to FLT3L (29). We used this system to examine IRF protein levels in BM-derived DC. FLT3L-derived pDC expressed high levels of IRF7, as well as IRF3 (Fig. 3B, upper panels). In contrast, GM-CSF-derived mDC expressed IRF3 but markedly less IRF7. Naive BM progenitors expressed undetectable levels of either IRF protein (Fig. 3B, upper panels, first lane).
In vitro differentiation of DC in response to FLT3L yielded heterogeneous populations, with typically 30–40% being CD11b−CD11cint. Wright-Giemsa staining of cytospin preparations confirmed that the major cell population displayed plasmacytoid morphology (Fig. 3C) and were B220+ (data not shown). In contrast, GM-CSF treatment resulted in the generation of a DC population that was 75–90% CD11b+CD11c+B220− and resembled DC with a myeloid morphology (Fig. 3C). The addition of interleukin-3 to differentiating bone marrow cultures, either alone or in the presence of FLT3L or GM-CSF, skewed cell differentiation away from DC and yielded cultures dominated by granulocyte/macrophage lineages that did not express IRF7 and expressed little IRF3 (Fig. 3B and C, and data not shown). IFN has been suggested to bias DC differentiation toward pDC morphology (30), but we observed no such effect when IFNα was added alone or in the presence of FLT3L (Fig. 3B and data not shown). However, we found that inclusion of stem cell factor increased the yield of FLT3-derived pDC, probably by enhancing progenitor proliferation during the first few days of culture (data not shown).
Similarly, high levels of IRF7 were detected in splenic pDC populations isolated by FACS from spleens of uninfected mice (Fig. 3D). These CD11cintLy6hiB220+ cells expressed much higher levels of IRF7 than the moderate levels of IRF7 expressed in a mixed DC population isolated by sorting splenocytes only on the basis of CD11c or the minimal levels of IRF7 expressed by in vitro differentiated mDC. Taken together, these results indicate that the major if not exclusive cells expressing constitutive IRF7 are pDC.
Bone Marrow-derived pDC Produce IFNα in Response to Viral Infection
In vitro differentiated pDC and mDC populations were infected with NDV and assayed for levels of IRF7 protein and IFNα mRNA. FLT3L-differentiated pDC populations expressed IRF7 protein prior to infection, and the level of IRF7 increased modestly following infection (Fig. 3B, lower panels). IRF7 protein was nearly undetectable in naive mDC, but it was inducible, although to relatively lower levels, following viral infection. Consistent with the higher constitutive levels of IRF7, infected pDC produced ~10-fold more IFNα (all subtypes) than did mDC. These cells expressed ~8-fold more of the IRF7-dependent non-α4 subtypes of IFNα than did mDC, and expression peaked more rapidly, at 4 h p.i. (Fig. 4A and data not shown). These amounts may underestimate the potential of pDC to produce IFNα since these in vitro derived cultures were only ~30–40% pDC by flow cytometric and histologic criteria (data not shown). These results demonstrate that IRF7 is highly expressed in IFN-producing pDC in a largely IFN-independent manner, enabling rapid and robust IFN production in response to infection. Early IFN production included production of IRF7-dependent non-α4 subtypes, normally expressed only with delayed kinetics in other cell types.
Fig. 4. IFNα expression by DC correlates with IRF7 levels.
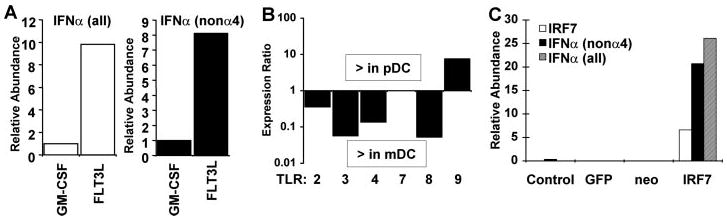
A, BM-derived DC cultured with GM-CSF (mDC) or FLT3L (pDC) were infected with NDV for 4 h, and mRNA levels of IFNα (all subtypes) (left) and IFNα (non-α4 subtypes) (right) were measured by quantitative real-time RT-PCR. B, relative expression levels of TLR species in BM-derived DC were measured by quantitative real-time RT-PCR. Data are shown as the ratio of expression in pDC relative to expression in mDC. Values above the abscissa represent greater expression in pDC, and values below the abscissa represent greater expression in mDC. C, 3T3 fibroblasts were either mock-infected or infected with retroviruses expressing green fluorescent protein (GFP), no product, or IRF7, as indicated. Two days later, cells were infected with NDV, and relative expression levels of IRF7 and IFNα (all subtypes and non-α4 subtype) were measured 4 h p.i. by quantitative real-time RT-PCR and normalized to ribosomal protein L32 mRNA. The data are representative of three experiments.
Differential Expression of TLR Proteins by Mouse DC Subsets
The ability of DC to respond to pathogens is determined at least in part by TLR proteins, although the role of these receptors in viral infections is controversial (31). We examined the levels of TLR mRNA expression in pDC and mDC. Interestingly, TLR2, TLR3, TLR4, and TLR8 were expressed at significantly higher levels in mDC than in pDC. Only TLR9 was expressed at a higher level in pDC populations (Fig. 4B), consistent with other reports (reviewed in Refs. 32–34). Since TLR9 is a DNA receptor, these results suggest that the enhanced IFN production by pDC cannot be explained by the presence of TLR proteins traditionally associated with ligands that could be derived from RNA viruses. Instead, IFN production by pDC more closely correlates with IRF7 abundance (Fig. 3). IRF8 was also modestly enriched in pDC (Fig. 3 and data not shown) and may contribute to the IFN-producing ability of these cells. However, IRF8 is required for DC differentiation (35, 36) and likely contributes to DC lineage commitment rather than directly to function.
Ectopic Expression of IRF7 Augments the Kinetics, Magnitude, and Diversity of IFNα Production
We designed an experiment to test whether IRF7 constitutive expression was a major determinant for the robust IFN production observed in pDC. Fibroblasts lack IRF7 expression prior to IFN exposure and therefore produce IFN in a delayed-early manner following viral infection, especially the non-α4 subset (4). We ectopically expressed IRF7 (or green fluorescent protein, as control) in mouse fibroblasts and measured IFN production following infection with NDV. As shown in Fig. 4C, IRF7-expressing fibroblasts responded to infection by making 100-fold more IFNα than control cells and produced it more rapidly, with significant levels detectable at 4 h p.i. The IFN produced included multiple subtypes and was not limited to the IFNα4 subtype produced in the absence of positive feedback. Thus, although additional components of the IFN signaling pathway are also induced during positive feedback, IRF7 expression alone is sufficient to drive the rapid production of large quantities of multiple isoforms of type I IFN following virus infection.
DISCUSSION
IFN production is a rapid and near universal response to viral infection. Previous in vitro studies documented a positive feedback model for robust IFN production, involving augmented responses due to induction of signaling components in response to limited early IFN production. Most cells are capable of mounting a modest response to viral infection by producing limited quantities of IFNβ and IFNα4. This response utilizes preexisting cellular components, but augmented production of an expanded repertoire of IFNα species requires feedback-dependent induction of additional components (4). The major induced component implicated in positive feedback is IRF7, although the contribution of other inducible components remains a possibility. Although this model has been widely documented in cell culture, its relevance in vivo has remained unclear. The results reported here clearly demonstrate a role for positive feedback in vivo during respiratory viral infections. Intranasal infection of mice with influenza virus resulted in robust IFN production only in mice capable of responding to IFN and mounting a positive feedback response (Fig. 1A). In these mice, production of IFN correlated with induced expression of IRF7.
In addition to a positive feedback response in infected lung, we found evidence of positive feedback in numerous tissues, at least at the level of IRF7 induction. Thus, in most tissues examined, IRF7 protein was constitutively expressed at relatively low to undetectable levels in the absence of infection. Only by following IFN production in vivo were significant amounts of IRF7 observed in such tissues as heart and liver. These results suggest that positive feedback is likely to play a significant role in IFN production, at least at local sites of infection.
A distinct pattern of IRF7 and IFN production was observed in hematopoietic organs. Spleen and thymus produced high constitutive amounts of IRF7, correlating with a rapid and robust production of IFN following viral infection. In contrast, fibroblasts were capable of producing levels of IFN similar to those produced by splenocytes only at late times after infection, after autocrine IFN had induced significant amounts of IRF7 protein. Subset analysis of splenocytes and in vitro differentiation of hematopoietic progenitors demonstrated that the vast majority of constitutive IRF7 was produced by pDC, the cell type associated with systemic IFN production. In contrast, other dendritic cell types, such as mDC, expressed relatively little IRF7 and were dependent on positive feedback for robust IFN production. Given the low but inducible expression of IRF7 in other tissues in vivo, we would predict them to produce IFN similarly to fibroblasts, subject to positive feedback. Remarkably, fibroblasts engineered to express high constitutive amounts of IRF7 acquired the ability to produce IFN in a manner that was at least superficially similar to pDC. These cells produced substantial levels of multiple IFNα species as early as 4 h p.i., a time point when control fibroblasts produced limited levels of a narrow spectrum of IFN species.
Kalinke and colleagues (14) have reached a different conclusion, questioning the significance of IRF7 and positive feedback for IFN production in vivo. They observed robust IFN production in infected IFNAR-deficient mice that cannot respond to IFN and concluded that positive feedback was not significant in vivo. However, their model examined IFN production following intravenous inoculation of virus, a route that likely directs the virus rapidly to the spleen. As we show here, the spleen is one organ that would not be expected to display a major component of positive feedback for IFN production, given the presence of resident pDC constitutively expressing IRF7. Indeed, they also observed that pDC-like cells were largely responsible for the IFN produced in response to intravenous viral inoculation, and they also observed a modest increase in IFN production over time that was limited to wild type mice, consistent with positive feedback.
As we show here, other routes of infection that do not result in systemic viremia result in a different outcome that is IRF7-dependent. Moreover, host responses to viruses that do not stimulate IFN production from pDC, such as lymphocytic choriomeningitis virus, are completely dependent on positive feedback (20). In contrast to our results, Kalinke and colleagues (14) did not detect a correlation between IRF7 mRNA levels and IFN production and concluded that IRF7 was not important for IFN produced by splenocytes. Such a conclusion is inconsistent with the data presented here but may have resulted from detecting IRF7 by the relatively insensitive in situ hybridization method rather than with the specific antibodies and quantitative real-time RT-PCR assays used here. It is highly likely that the fundamental mechanism of IFN transcription is the same in pDC as in other cells (i.e. IRF-dependent), the difference being that IRF7 is constitutively present. Constitutive expression of IRF7 protein in pDC also allows these cells to induce IFN in response to TLR7 and -9 engagement, a process that has recently been shown to signal directly through IRF7 (18, 19).
Recently, similar observations of constitutive IRF7 expression have been reported for human blood pDC (37–39). Although these reports did not examine IRF7 protein levels or a correlation with IFN kinetics and could not perform the systematic investigation of IRF7 expression in lymphoid organ subsets that we carried out in the mouse, they suggest that the patterns of IRF7 and IFN production we observed may be a general property of this subset of dendritic cells. From our results, there appear to be two modes of IFN production in vivo that differ in both temporal and quantitative aspects. One mode involves positive feedback induction of IRF7 that likely occurs in most peripheral organs, resulting in a relatively delayed response. A second mode relies on preformed IRF7 that is independent of positive feedback, but this is restricted to lymphoid organs due to the corresponding prevalence of resident pDC and results in a strong and immediate response.
A dual mode of IFN production as described here may provide distinct survival advantages for the host. A requirement for positive feedback for robust IFN production at local sites of infection may limit potentially destructive inflammatory responses that would occur in response to high levels of IFN production. Should initial IFN production be sufficient to clear infections, only modest levels of IFN would be produced since induction of IRF7 would be without effect in the absence of continued viral infection required to stimulate its phosphorylation. In contrast, systemic viremia would require a rapid and effective response that might best be accomplished by a specialized cell, such as the interferon-producing cell. Constitutive IRF7 expression in pDC could facilitate an immediate accelerated response to systemic viral infections, providing a survival advantage in the context of potentially overwhelming viremia.
IRF7 expression in pDC is quite remarkable, considering that the protein is readily detectable in whole spleen but is largely restricted to a subset of DC that together comprises a small fraction of the total splenic cellularity. IRF7 accumulation in spleen is largely equivalent in wild type and STAT1-deficient organs and therefore independent of IFN signaling. High constitutive splenic IRF7 was detected at both the mRNA and the protein level. Moreover, protein half-life measurements did not reveal any evidence that differential stability contributed to this accumulation.2 These findings suggest that IRF7 is regulated at the transcriptional level, implying a distinct transcriptional mechanism in pDC relative to other cell types, including the closely related mDC. Interestingly, a similar conclusion has recently been reached for the transcription factor CIITA that is required for major histocompatibility complex class II expression. CIITA expression was found to depend on a distinct promoter in pDC relative to conventional DC and most other cell types (40). It will be of interest to determine whether IRF7 is similarly regulated through a pDC-specific transcriptional mechanism that enables high constitutive expression in this cell type.
Acknowledgments
We thank Kimberly O'Malley for assistance with experiments and unpublished data, Heidi Cleven and Tom Moran for advice on GM-CSF bone marrow cultures, Sandra Diebold and Caetano Reis e Sousa for advice on pDC isolation and for providing comparative samples, Gary Nolan for reagents, and Ramon Gimeno, Isabelle Marie, Yaming Wang, and other members of the laboratory for assistance, advice, and comments on the manuscript.
Footnotes
This work was supported by National Institutes of Health grants from the Public Health Service and by a grant-in-aid from the American Heart Association. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The abbreviations used are: IFN, interferon; IFNAR, IFN-α receptor; IRF, IFN regulatory factor; ISG, IFN-stimulated genes; ISGF3, ISG factor 3; DC, dendritic cell(s); m, myeloid; p, plasmacytoid; TLR, toll-like receptor; BM, bone marrow; RT, reverse transcriptase; NDV, New-castle disease virus; p.i., post-infection; FLT3L, Fms-like tyrosine kinase 3 ligand; GM-CSF, granulocyte-macrophage colony-stimulating factor; STAT, signal transducers and activators of transcription; FACS, fluorescence-activated cell sorter.
References
- 1.Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD. Annu Rev Biochem. 1998;67:227–264. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- 2.Levy DE, Marie I, Prakash A. Curr Opin Immunol. 2003;15:52–58. doi: 10.1016/s0952-7915(02)00011-0. [DOI] [PubMed] [Google Scholar]
- 3.Yoneyama M, Suhara W, Fukuhara Y, Fukuda M, Nishida E, Fujita T. EMBO J. 1998;17:1087–1095. doi: 10.1093/emboj/17.4.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marié I, Durbin JE, Levy DE. EMBO J. 1998;17:6660–6669. doi: 10.1093/emboj/17.22.6660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sato M, Hata N, Asagiri M, Nakaya T, Taniguchi T, Tanaka N. FEBS Lett. 1998;441:106–110. doi: 10.1016/s0014-5793(98)01514-2. [DOI] [PubMed] [Google Scholar]
- 6.Sharma S, tenOever BR, Grandvaux N, Zhou GP, Lin R, Hiscott J. Science. 2003;300:1148–1151. doi: 10.1126/science.1081315. [DOI] [PubMed] [Google Scholar]
- 7.Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao SM, Maniatis T. Nat Immunol. 2003;4:491–496. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- 8.Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, maizumi T, Miyagishi M, Taira K, Akira S, Fujita T. Nat Immunol. 2004;5:730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- 9.Andrejeva J, Childs KS, Young DF, Carlos TS, Stock N, Goodbourn S, Randall RE. Proc Natl Acad Sci U S A. 2004;101:17264–17269. doi: 10.1073/pnas.0407639101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Durbin JE, Hackenmiller R, Simon MC, Levy DE. Cell. 1996;84:443–450. doi: 10.1016/s0092-8674(00)81289-1. [DOI] [PubMed] [Google Scholar]
- 11.Meraz MA, White JM, Sheehan KC, Bach EA, Rodig SJ, Dighe AS, Kaplan DH, Riley JK, Greenlund AC, Campbell D, Carver-Moore K, DuBois RN, Clark R, Aguet M, Schreiber RD. Cell. 1996;84:431–442. doi: 10.1016/s0092-8674(00)81288-x. [DOI] [PubMed] [Google Scholar]
- 12.Durbin JE, Fernandez-Sesma A, Lee CK, Rao TD, Frey AB, Moran TM, Vukmanovic S, Garcia-Sastre A, Levy DE. J Immunol. 2000;164:4220–4228. doi: 10.4049/jimmunol.164.8.4220. [DOI] [PubMed] [Google Scholar]
- 13.Garcia-Sastre A, Durbin RK, Zheng H, Palese P, Gertner R, Levy DE, Durbin JE. J Virol. 1998;72:8550–8558. doi: 10.1128/jvi.72.11.8550-8558.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barchet W, Cella M, Odermatt B, Asselin-Paturel C, Colonna M, Kalinke U. J Exp Med. 2002;195:507–516. doi: 10.1084/jem.20011666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Diebold SS, Montoya M, Unger H, Alexopoulou L, Roy P, Haswell LE, Al-Shamkhani A, Flavell R, Borrow P, Reis e Sousa C. Nature. 2003;424:324–328. doi: 10.1038/nature01783. [DOI] [PubMed] [Google Scholar]
- 16.Asselin-Paturel C, Boonstra A, Dalod M, Durand I, Yessaad N, Dezutter-Dambuyant C, Vicari A, O’Garra A, Biron C, Briere F, Trinchieri G. Nat Immunol. 2001;2:1144–1150. doi: 10.1038/ni736. [DOI] [PubMed] [Google Scholar]
- 17.Boonstra A, Asselin-Paturel C, Gilliet M, Crain C, Trinchieri G, Liu YJ, O’Garra A. J Exp Med. 2003;197:101–109. doi: 10.1084/jem.20021908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Honda K, Yanai H, Mizutani T, Negishi H, Shimada N, Suzuki N, Ohba Y, Takaoka A, Yeh WC, Taniguchi T. Proc Natl Acad Sci U S A. 2004;101:15416–15421. doi: 10.1073/pnas.0406933101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kawai T, Sato S, shii KJ, Coban C, Hemmi H, Yamamoto M, Terai K, Matsuda M, noue JI, Uematsu S, Takeuchi O, Akira S. Nat Immunol. 2004;5:1061–1068. doi: 10.1038/ni1118. [DOI] [PubMed] [Google Scholar]
- 20.Dalod M, Salazar-Mather TP, Malmgaard L, Lewis C, Asselin-Paturel C, Briere F, Trinchieri G, Biron CA. J Exp Med. 2002;195:517–528. doi: 10.1084/jem.20011672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dalod M, Hamilton T, Salomon R, Salazar-Mather TP, Henry SC, Hamilton JD, Biron CA. J Exp Med. 2003;197:885–898. doi: 10.1084/jem.20021522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Levy DE. J Exp Med. 2002;195:F15–18. doi: 10.1084/jem.20020075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Inaba K, naba M, Romani N, Aya H, Deguchi M, kehara S, Muramatsu S, Steinman RM. J Exp Med. 1992;176:1693–1702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Au WC, Moore PA, LaFleur DW, Tombal B, Pitha PM. J Biol Chem. 1998;273:29210–29217. doi: 10.1074/jbc.273.44.29210. [DOI] [PubMed] [Google Scholar]
- 25.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 26.Tovey MG, Content J, Gresser I, Gugenheim J, Blanchard B, Guymarho J, Poupart P, Gigou M, Shaw A, Fiers W. J Immunol. 1988;141:3106–3110. [PubMed] [Google Scholar]
- 27.Hata N, Sato M, Takaoka A, Asagiri M, Tanaka N, Taniguchi T. Biochem Biophys Res Commun. 2001;285:518–525. doi: 10.1006/bbrc.2001.5159. [DOI] [PubMed] [Google Scholar]
- 28.Lee CK, Gimeno R, Levy DE. J Exp Med. 1999;190:1451–1464. doi: 10.1084/jem.190.10.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gilliet M, Boonstra A, Paturel C, Antonenko S, Xu XL, Trinchieri G, O’Garra A, Liu YJ. J Exp Med. 2002;195:953–958. doi: 10.1084/jem.20020045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Honda K, Sakaguchi S, Nakajima C, Watanabe A, Yanai H, Matsumoto M, Ohteki T, Kaisho T, Takaoka A, Akira S, Seya T, Taniguchi T. Proc Natl Acad Sci U S A. 2003;100:10872–10877. doi: 10.1073/pnas.1934678100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Levy DE, Marie IJ. Nat Immunol. 2004;5:699–701. doi: 10.1038/ni0704-699. [DOI] [PubMed] [Google Scholar]
- 32.Barton GM, Medzhitov R. Curr Top Microbiol Immunol. 2002;270:81–92. doi: 10.1007/978-3-642-59430-4_5. [DOI] [PubMed] [Google Scholar]
- 33.Barton GM, Medzhitov R. Science. 2003;300:1524–1525. doi: 10.1126/science.1085536. [DOI] [PubMed] [Google Scholar]
- 34.Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. Science. 2004;303:1529–1531. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- 35.Tsujimura H, Tamura T, Ozato K. J Immunol. 2003;170:1131–1135. doi: 10.4049/jimmunol.170.3.1131. [DOI] [PubMed] [Google Scholar]
- 36.Tsujimura H, Tamura T, Gongora C, Aliberti J, Reis e Sousa C, Sher A, Ozato K. Blood. 2003;101:961–969. doi: 10.1182/blood-2002-05-1327. [DOI] [PubMed] [Google Scholar]
- 37.Dai J, Megjugorac NJ, Amrute SB, Fitzgerald-Bocarsly P. J Immunol. 2004;173:1535–1548. doi: 10.4049/jimmunol.173.3.1535. [DOI] [PubMed] [Google Scholar]
- 38.Kerkmann M, Rothenfusser S, Hornung V, Towarowski A, Wagner M, Sarris A, Giese T, Endres S, Hartmann G. J Immunol. 2003;170:4465–4474. doi: 10.4049/jimmunol.170.9.4465. [DOI] [PubMed] [Google Scholar]
- 39.Izaguirre A, Barnes BJ, Amrute S, Yeow WS, Megjugorac N, Dai J, Feng D, Chung E, Pitha PM, Fitzgerald-Bocarsly P. J Leukocyte Biol. 2003;74:1125–1138. doi: 10.1189/jlb.0603255. [DOI] [PubMed] [Google Scholar]
- 40.LeibundGut-Landmann S, Waldburger JM, Reis e Sousa C, Acha-Orbea H, Reith W. Nat Immunol. 2004;5:899–908. doi: 10.1038/ni1109. [DOI] [PubMed] [Google Scholar]